INTRODUCTION
Bone is a crucial tissue that provides internal skeletal support for every organ, as well as forming and structuring the entire human frame. Bone homeostasis is delicately regulated by osteoclast-mediated bone resorption and osteoblast-induced bone formation.[1] Disruption of this balance perturbs steady-state bone remodeling. This leads to severe bone diseases, including osteoporosis and osteopenia, that are directly associated with excessive bone destruction and impaired bone quality.[2] Therefore, it would be useful to target osteoclasts and/or osteoblasts in the development of new drugs against bone-destructive diseases.
Osteoclasts, derived from hematopoietic precursors of the monocyte/macrophage lineage, are bone-specialized multinucleated cells. The differentiation of osteoclasts depends mainly on two critical cytokines, receptor activator of nuclear factor-kappa B (NF-kB) ligand (RANKL) and macrophage colony-stimulating factor (M-CSF), which are supplied by stromal cells or osteoblasts.[3,4] M-CSF is important for the proliferation and survival of the osteoclast precursors and upregulates RANK expression, which is a prerequisite for osteoclastogenesis.[5] RANKL, a member of the tumor necrosis factor (TNF) family, plays crucial roles in osteoclast differentiation and activation.[3,6] The binding of RANKL to its receptor RANK in osteoclast precursor cells induces TNF receptor-associated factor 6 (TRAF6) recruitment and the sequential activation of multiple intracellular signaling pathways including the mitogen-activated protein kinase (MAPK) pathways extracellular signal-regulated kinase (ERK), c-Jun-N-terminal kinase (JNK), and p38 MAPK, and the NF-kB pathway.[7,8,9] These signaling cascades lead to the induction and activation of osteoclastogenic transcription factors such as c-Fos and nuclear factor of activated T cells (NFATc1).[10,11,12] NFATc1 then translocates to the nucleus and activates the expression of multiple osteoclastogenesis-related genes, such as cathepsin K (CTK), c-Src, and tartrate-resistant acid phosphatase (TRAP).[13]
Recently, many plants and their extracts have been recognized as useful sources for the prevention and treatment of bone-related disorders. Species in the genus Euphorbia belong to the Euphorbiaceae family, which has a long history of use as medicinal plants in traditional treatments.[14] Some of their components have shown considerable potential as lead compounds in drug discovery and have been the focus of many medicinal chemistry and therapeutic studies.[14] Euphorbia lathyris L. (ELL) is native to southern Europe, Africa, and Asia. As part of a search for bioactive compounds, we investigated the effect of the methanol extract of the aerial part of ELL on RANKL-induced osteoclastogenesis using mouse primary osteoclast precursors. Our results suggested that ELL has potential as a natural herbal therapy for diseases associated with bone loss.
METHODS
1. Reagents
The methanol extract of ELL was obtained from the National Institute of Horticultural and Herbal Science. Briefly, the aerial portion of ELL was extracted in 99.99% methyl alcohol at 50℃ using an accelerated solvent extractor (ASE). After filtration, the extract was concentrated using a rotary evaporator (JP-SD1000; Eyela, Tokyo Rikikatai, Japan). The final extract was dissolved in dimethyl sulfoxide (Sigma Aldrich, St. Louis, MO, USA) and then diluted in phosphate buffered saline (PBS). Antibodies against ERK, phospho-ERK, phospho-p38, p38, inhibitor of kappa B (IκB), β-actin, and c-Fos were purchased from Cell Signaling Technology (Beverly, MA, USA). The antibody against NFATc1 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA).
2. Co-culture system
Primary calvarial osteoblasts were obtained from the calvariae of neonatal ICR mice (Samtako Inc., Osan, Korea) as previously described.[15] Bone marrow cells were obtained from the long bones of 4- to 6-week-old ICR male mice. To examine osteoclast formation, mouse bone marrow cells (1×105 cells) were co-cultured with calvarial osteoblasts (5×103 cells) with 1,25-dihydroxy-vitamin D3 (1,25-[OH]2D3, 10 nM) in the presence or absence of the methanol extract in 96-well culture plates (Corning Life Sciences, Acton, MA, USA). After six days of culture, the cells were fixed, then permeabilized with a mixture of acetone and ethanol (1:1 volume ratio) for 30 sec, and treated with TRAP staining solution (0.01% naphthol AS-MX phosphate [Sigma-Aldrich] and 0.06% Fast Red Violet LB Salt [Sigma-Aldrich] in 50 mM sodium tartrate dehydrate and 45 mM sodium acetate, at pH 5.0). TRAP-positive (TRAP+) multinucleated cells (>5 nuclei/ cell) were considered mature osteoclasts.
3. BMM culture system
Bone marrow cells were obtained from the long bones of 8- to 10-week-old ICR mice (Samtako Inc.). Bone marrow cells were cultured in the presence of M-CSF (30 ng/mL; PeproTech Inc., Rocky Hill, NJ, USA) for three days to generate the bone marrow-derived macrophages (BMMs). To examine osteoclast formation, BMMs were treated with the methanol extract of ELL in the presence of M-CSF (30 ng/mL) and RANKL (100 ng/mL; PeproTech Inc.) in 96-well culture plates (Corning Life Sciences). After four days, the cells were fixed and stained for TRAP.
4. Cell cytotoxicity assay
Cell cytotoxicity was determined by the microtitration (MTT) assay. BMMs (1×104 cells/well) were placed in a 96-well plate and cultured with M-CSF (30 ng/mL, R & D) and the methanol extract of ELL in alpha- minimum essential medium (α-MEM) for 48 hr. The MTT solution was added and incubated in the dark. After 5 hr, solubilization buffer (10% sodium dodecyl sulfate [SDS] in 0.01 M hydrochloric acid [HCl]) was added and the cells were incubated overnight. The proliferation of the BMMs was determined by measuring the optical density (OD) of the wells at 570 nm.
5. RNA extraction and polymerase chain reaction (PCR) assay
Total RNA was purified with Easy-Blue (iNtRON Biotechnology Inc., Seongnam, Korea). The cDNA was synthesized from 5 µg of RNA by using the Revert Aid™ first-strand cDNA synthesis kit (iNtRON Biotechnology Inc.) and amplified using real-time PCR or reverse transcription (RT)-PCR. The following primers of osteoclastogenic genes were used in this study: RANKL: 5′-CCA AGA TCT CTA ACA TGA CG-3′ (forward), 5′-CAC CAT CAG CTG AAG ATA GT-3′ (reverse); RANK: 5′-CGA GGA AGA TTC CCA CAG AG-3′ (forward), 5′-CAG TGA AGT CAC AGC CCT CA-3′ (reverse); osteoprotegerin (OPG): 5′-ACG GAC AGC TGG CAC ACC AG-3′ (forward), 5′-CTC ACA CAC TCG GTT GTG GG-3′ (reverse); calcitonin receptor (CTR): 5′-TTT CAA GAA CCT TAG CTG CCA GAG-3′ (forward), 5′-CAA GGC ACG GAC AAT GTT GAG AAG-3′ (reverse); CTK: 5′-CTT CCA ATA CGT GCA GCA GA-3′ (forward), 5′-ACG CAC CAA TAT CTT GCA CC-3′ (reverse); GAPDH: 5′-AAC GGA TTT GGT CGT ATT GGG-3′(forward), 5′-CAG GGG TGC TAA GCA GTT GG-3′ (reverse); β-actin, 5′-TTT GAT GTC ACG CAC GAT TTC C-3′ (forward), 5′-TGT GAT GGT GGG AAT GGG TCA G-3′ (reverse). Real-time PCR reactions for RANK, RANKL and OPG were performed in a total volume of 20 µL using SYBR® Green CTR Master Mix (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. Thermocycling was performed using a 7500 Real-time PCR System (Applied Biosystems) with the following conditions: initial hold, 95℃ for 10 min; followed by 40 cycles of denaturation at 95℃ for 15 sec, annealing at 58℃, and extension at 60℃ for 1 min. An index mRNA level was assessed using a threshold cycle value and normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. The RT-PCR conditions for CTR, CTK, and β-actin were as follows: an initial denaturation step at 94℃ for 3 min, followed by 28 cycles (CTR) or 22 cycles (CTK, β-actin) of denaturation at 94℃ for 30 sec, annealing at 58℃ for 45 sec, and extension at 72℃ for 60 sec; and a final extension at 72℃ for 10 min. After amplification, the RT-PCR reaction mixtures were electrophoresed on 1% agarose gel and visualized by ethidium bromide staining and ultraviolet (UV) irradiation.
6. Western blot analysis
Total cell lysates were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto Immobilon-P membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% non-fat-milk in PBS-Tween (PBS-T), immunostained with anti-phospho ERK (1:1,000), anti-phospho p38 (1:1,000), anti-ERK (1:1,000), anti-p38 (1:1,000), anti-IκB (1:1,000), anti-NFATc1 (1:200), anti-c-Fos (1:1,000), and anti-β-actin (1:4,000), and incubated with a horseradish peroxidase-conjugated secondary antibody (1:5,000). The membranes were developed using an advanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Buckinghamshire, UK).
7. Bone resorption assay
BMMs were differentiated on dentine slices with M-CSF (30 ng/mL) and RANKL (100 ng/mL) for four days, and then treated with the methanol extract of ELL for two days. The surfaces of the dentine slices were wiped to remove the cells, and the slices were then stained with toluidine blue (1 µg/mL) (J.T. Baker, Phillipsburg, NJ, USA). The number of pits formed by bone resorption on the dentine slices was counted.
RESULTS
1. ELL suppressed RANKL-induced osteoclast formation and bone resorption
First, we examined the effect of ELL on RANKL-induced osteoclast differentiation using mouse BMMs. BMMs were cultured with RANKL and M-CSF in the presence or absence of the methanol extract of ELL for four days. BMMs differentiated into mature TRAP-positive (TRAP+) multinucleated osteoclasts (MNCs) during the culture period, while treatment with ELL inhibited osteoclast differentiation in a dose-dependent manner (Fig. 1A). The MTT assay showed the anti-osteoclastogenic effect of ELL was not attributable to cellular toxicity (Fig. 1B). Osteoclast differentiation is associated with the upregulation of specific genes in response to RANKL.[8] We further assessed the inhibitory effect of ELL on osteoclastogenesis by evaluating the RANKL-induced mRNA expression levels of osteoclast-related genes such as CTR and CTK. In the RT-PCR analysis, the expression levels of those genes were dramatically increased in BMMs challenged by RANKL and M-CSF for four days. However, treatment with ELL significantly inhibited RANKL-induced osteoclast-related gene expression (Fig. 1C). We further demonstrated that the mRNA expression level of RANK was significantly decreased in BMMs by the treatment of ELL (Fig. 1D). Bone resorption activity occurs after the terminal maturation and activation of osteoclasts. To examine whether the effect of ELL on osteoclast formation could be reflected in osteoclastic activity, we performed an in vitro resorption pit assay using a dentine slice. Many resorption pits were generated with RANKL-treated cells (Fig. 1E). In contrast, the treatment of ELL strongly inhibited the formation of resorption pits by the RANKL-treated cells. Therefore, these results suggested that the methanol extract of ELL exerted inhibitory effects on osteoclast formation which led to reduced bone resorption.
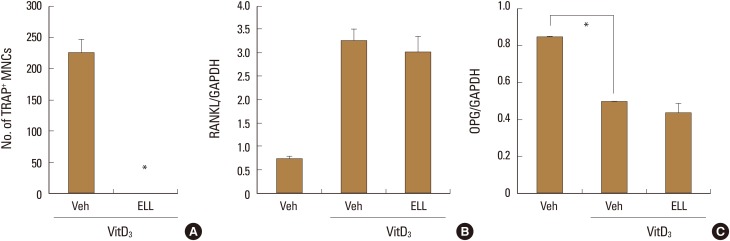
2. ELL did not modulate mRNA expression of RANKL and OPG in osteoblasts
It is known that osteoblasts express RANKL in response to stimulation by bone-resorbing factors such as 1α,25-(OH)2D3. [1] Thus in co-cultures of mouse primary osteoblasts and bone marrow cells, osteoclasts are formed in the presence of 1α,25-(OH)2D3.[15] To investigate whether ELL effects the ability of osteoblasts to support osteoclast differentiation, we added ELL to co-cultures of mouse primary osteoblasts and bone marrow cells. ELL inhibited the differentiation of bone marrow cells into osteoclasts induced by 1α, 25-(OH)2D3 at 50 µM (Fig. 2A). To clarify whether the expression level of RANKL was involved in the inhibitory effect of ELL on osteoclast formation, we examined the effects of ELL on the mRNA expression level of RANKL. Consistent with a previous report,[16] 1α,25-(OH)2D3 induced the mRNA expression of RANKL in primary osteoblasts, but ELL showed little effect on it (Fig. 2B). Osteoblasts are also known to produce a soluble decoy receptor for RANKL, called OPG, which inhibits osteoclast formation by interrupting the interaction between RANKL and RANK.[17] When we further investigated the effect of ELL on the mRNA expression of OPG, ELL had no effect on the level of OPG mRNA suppressed by 1α,25-(OH)2D3 (Fig. 2C). These results suggest that ELL inhibits osteoclast formation by direct action on osteoclast precursors, and not via indirect action on osteoblasts.
3. ELL downregulated RANKL-induced expression of NFATc1 and c-Fos via the p38 MAPK pathway
As it has been suggested that osteoclast precursors may respond to ELL during osteoclastogenesis, we examined the molecular mechanism of the inhibitory effect of ELL using BMMs. We first investigated the effects of ELL on NFATc1 and c-Fos expression levels. It has been reported that NFATc1 is upregulated by RANKL and is important for osteoclast differentiation.[13] Furthermore, c-Fos is known to bind to and regulate the expression of NFATc1.[8,10] As reported previously, RANKL stimulation increased the expression of c-Fos and NFATc1 in BMMs. ELL abolished the RANKL-induced protein expression of c-Fos and NFATc1 (Fig. 3A, B).
RANKL stimulates several signaling pathways that involve MAPKs (ERK, JNK, and p38), and NF-κB.[8,18,19] When we examined the effects of ELL on the early signaling pathways induced by RANKL in BMMs, we found that ELL pretreatment suppressed the phosphorylation by RANKL of p38, but not ERK (Fig. 3C, D). We also examined effects on the NF-κB signaling pathway. Upon RANKL stimulation, NF-κB was activated through IκB-kinase (IKK) activation and the subsequent phosphorylation and degradation of IκB.[20] RANKL stimulation led to the degradation of IκB within 15 min, although ELL failed to interfere with this process (Fig. 3E). Altogether, the results suggested that ELL downregulated RANKL-induced expression of NFATc1 and c-Fos via the p38 MAPK pathway.
DISCUSSION
Bone homeostasis is a balance of osteoblast-mediated bone formation and osteoclast-mediated bone resorption.[1] Deficiency in bone formation by osteoblasts or excessive bone resorption by osteoclasts can result in metabolic bone disorders such as osteoporosis and rheumatoid arthritis. Drugs that either increase bone building or block bone degradation have been developed for the treatment of osteoporosis.[20] There is a growing interest in the efficacy of natural plant products for the prevention and treatment of bone-related disorders. In addition, traditional herbal medicine is a successful source of therapeutic agents and drug leads. Therefore, we screened several herbal extracts to discover potential therapeutic agents with anti-osteoclastogenic activity. Methanol has been used to make herbal preparations for long periods. Since methanol provides an effective way of maximizing the bioavailability of active components, we used methanol extracts of various herbs.
In this study, we have shown that ELL effectively inhibits RANKL-induced osteoclast differentiation without cytotoxicity. The suppressive effect of ELL on pit formation on dentine slices also suggested that ELL inhibits the bone resorptive function of osteoclasts.
Under pathophysiological conditions, RANKL-RANK signaling leads to the robust induction of NFATc1, which is a necessary factor for osteoclast differentiation.[21,22] For this pathway, the activation of the MAPK and NF-κB pathways is a prerequisite for osteoclast differentiation.[23,24] The addition of ELL to osteoclast precursors attenuated the RANKL-induced phosphorylation of p38. Furthermore, ELL also inhibited RANKL-induced NFATc1. NFATc1 is a key transcription factor for the expression of TRAP and other osteoclastogenesis-associated genes. The introduction of an NFATc1 siRNA converts TRAP+ cells into TRAP-negative cells. [25] In BMMs therefore, the inhibitory mechanism of ELL appears to be related to this pathway.
It has been well known that RANK requires a co-stimulatory signal to initiate osteoclastogenesis.[26] Although ELL supressed the mRNA expression level of RANK in BMMs, further study will be needed to elucidate the effect of ELL on the co-stimulatory signal.
In this study, we also demonstrated that ELL inhibits osteoclast formation in co-cultures of mouse primary osteoblasts and bone marrow cells. In mouse cell co-cultures, osteoclasts are formed in response to bone-resorbing factors such as 1α,25-(OH)2D3, parathyroid hormone (PTH), prostaglandin E2 (PGE2), and interleukin (IL)-11.[3] Three independent signals have been proposed to induce RANKL expression in osteoblasts: vitamin D receptor mediated signals induced by 1 α,25-(OH)2D3, cyclic adenosine 3',5'-monophosphate (cAMP)/protein kinase A (PKA)-mediated signals induced by PTH or PGE2, and gp130-mediated signals induced by IL-11. Of these different signals, vitamin D receptor- and cAMP/PKA-mediated signals suppress OPG expression in osteoblasts.[3] These pathways for RANKL gene expression could be one of the major targets of anti-resorptive agents. We further determined that ELL had little effect on the expression levels of RANKL and OPG mRNAs in osteoblasts treated with 1α,25-(OH)2D3, suggesting that ELL acted directly on osteoclast precursors. Although the precise inhibitory mechanism of ELL remains unidentified, we proposed that ELL might function as a potential therapeutic medication for attenuating osteoclast formation for the prevention and treatment of bone diseases such as osteoporosis.
In summary, the present data suggested that ELL has an inhibitory effect on osteoclast differentiation and function in vitro. The effectiveness of ELL on osteoclastogenesis suggests a potential application in therapeutic strategies which target the differentiation of osteoclasts in addition to their function. Consequently, continued advanced studies on the in vivo efficacy of ELL, as well as identification of the anti-osteoclastogenic components of ELL, will provide a basis for the development of new therapeutic agents.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print