Molecular and Cellular Crosstalk between Bone and Brain: Accessing Bidirectional Neural and Musculoskeletal Signaling during Aging and Disease
Article information

Abstract
Molecular omics technologies, including proteomics, have enabled the elucidation of key signaling pathways that mediate bidirectional communication between the brain and bone tissues. Here we provide a brief summary of the clinical and molecular evidence of the need to study the bone–brain axis of cross-tissue cellular communication. Clear clinical and molecular evidence suggests biological interactions and similarities between bone and brain cells. Here we review the current mass spectrometric techniques for studying brain and bone diseases with an emphasis on neurodegenerative diseases and osteoarthritis/osteoporosis, respectively. Further study of the bone–brain axis on a molecular level and evaluation of the role of proteins, neuropeptides, osteokines, and hormones in molecular pathways linked to bone and brain diseases is critically needed. The use of mass spectrometry and other omics technologies to analyze these cross-tissue signaling events and interactions will help us better understand disease progression and comorbidities and potentially identify new pathways and targets for therapeutic interventions. Proteomic measurements are particularly favorable for investigating the role of signaling and secreted and circulating analytes and identifying molecular and metabolic pathways implicated in age-related diseases.
INTRODUCTION
Mass spectrometric methodologies and depth of coverage in the human and other proteomes have rapidly and innovatively advanced in the past decades.[1] This had led to a dramatic expansion of the tissue types investigated and improved the accuracy and robustness of quantitative workflows. However, some biomedical research and selected tissues, specifically cross-tissue and cross-organ communication and interplay, remain greatly understudied. For example, proteomic characterization of skeletal tissue (e.g., bone, bone marrow, cartilage, and synovial fluid) lags behind other tissue types in spite of our expanding understanding of its role in systemic endocrine regulation. Proteomic studies have been limited in these tissues mainly due to the complex, dense, and mineralized bone matrix. The bone extracellular matrix (ECM) is rich in structural proteins including collagens, glycoproteins, and other abundant ECM proteins that present a challenge to the efficient extraction and detection of low-abundance signaling molecules relevant to biologic studies. Additionally, the mineralized ECM of bone contains large amounts of calcium in the form of matrix-bound hydroxyapatite, Ca10(PO4)6(OH)2, embedded within and around collagen fibrils. Nonspecific mineral interactions between charged amino acids, phosphate groups, and carboxyl residues often complicate conventional protein lysis extractions.[2] Prompted in part by interest in crosstalk among skeletal and other tissues, several attempts have been presented to further refine protein and material extraction protocols from bone, both from the native and de-mineralized bone matrix.
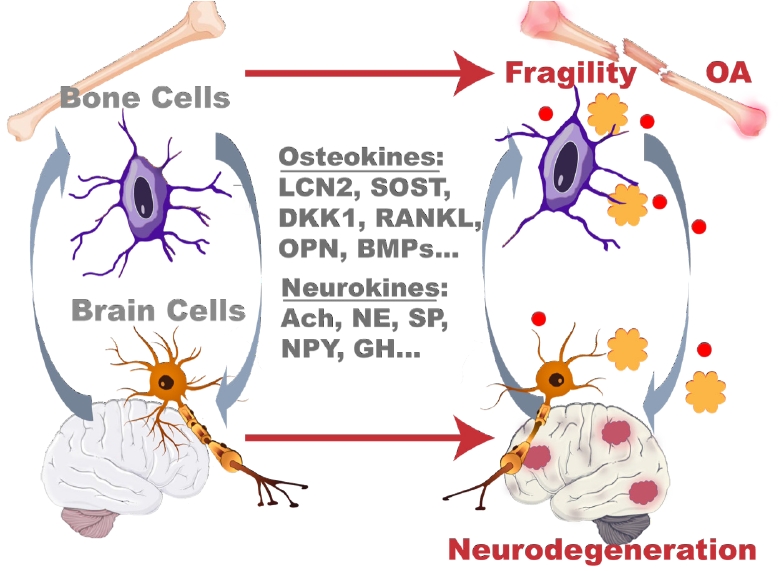
As the endocrine functions of bone and skeletal tissue have attracted more and more attention, the clinical evidence for the bidirectional molecular regulation between the brain and bone has emerged and become a focus for studies searching for new translatable therapies for diseases. These include Alzheimer’s disease (AD) and related dementias (ADRD), Parkinson’s disease (PD), osteoporosis, osteoarthritis (OA), and other neuro- and skeletal degenerative diseases. Striking similarities between neuronal and skeletal cells are highlighted by their shared cellular phenotypes, such as osteoclasts and microglia, which are distinct tissue-specific macrophages in the bone and brain, respectively. Interestingly, there are similarities in the network-like structures of both neurons and osteocytes. Given the rising need to study the bone-brain axis on a molecular level, we review clinical and molecular evidence for the biologic interaction and similarities between bone and brain cell types. We intend to motivate the field to use mass spectrometry proteomics to determine what role proteins, neuropeptides (NPs), hormones, and osteokines have in molecular pathways linked to bone and brain diseases.
The prevalence of both skeletal and neurologic disorders increases with age, and as individual health and lifespans increase across the world, the incidences of age-related complications are becoming the forefront of modern health research. The World Health Organization’s Global Burden of Disease Study, an epidemiological study in 21 regions worldwide, reported a 26.6% increase in knee OA from 1990 to 2010, while rates within the USA rose to 23% by 2017.[3,4] Despite advances in clinical prescreening and preventive care, rates of osteoporosis, clinically low bone mass linked to low-impact bone fracture, among adults aged 50+ marginally increased from 9.4% in 2007–2008 to 12.6% in 2017–2018,[5] with an estimated global burden for women as high as 23% in 2021.[6] Neurodegenerative diseases, such as AD and PD, follow similar trends with a 146% increase in deaths related to AD from 2000 to 2018 for a total of 122,000 deaths in the USA. The population burden of ADRD is predicted to double from 1.65% of adults over 65 to 3.3% by 2060 in the USA when 13.9 million Americans are projected to have the disease.[7] Worldwide, 78 million individuals are predicted to suffer from one form of dementia by 2030, with as many as 139 million by 2050 across all socioeconomic groups.[8] The prevalence and debilitating nature of these diseases is a rising health concern worldwide motivating the search for new avenues of research and treatment.
While skeletal and neurodegenerative diseases are comorbidities related to pathologic aging, their co-incidence has become a focus of age-related research. The focus on the “bone-brain axis” has been prompted by many newly realized paradigms. For example, the brain contributes to bone homeostasis and regeneration through the "efferent nervous system” in which the sympathetic nervous system regulates osteoblast and osteoclast differentiation, proliferation, and function through a host of secreted signaling factors.[9–11] This axis is bi-directional with several osteokines or bone-derived signaling factors, such as osteocyte-secreted inhibitors of the Wingless/Integrated-1 (Wnt) signaling pathway sclerostin (SOST) and dickkopf Wnt signaling pathway inhibitor 1 (DKK1) possibly able to cross the blood-brain barrier (BBB) and affect neural circuits [12,13] especially in age and disease states where the BBB is weakened or degraded.[14,15] These molecular interactions are strengthened by utilization of similar signaling mechanisms and pathways in each organ system and by the similarities of the cell types present within the nervous system and the skeleton. Thus, the susceptibility of both the bone and the brain to similar stimuli is an area of needed investigation to parse the bidirectionality of communication within this axis and the presence and consequences of these signaling effectors.
CLINICAL LINKS BETWEEN AGE-RELATED BONE AND BRAIN DISEASES
In addition to the histological observation of innervation of cortical bone,[16–19] the first hints of communication between the bone and brain come from clinical observations of comorbidities of skeletal and neurodegenerative conditions (Table 1). As early as the 1970s and 1980s, investigators began associating miscellaneous dementias and use of psychoactive drugs with increased fall risk in elderly patients more prone to bone fracture.[20,21] While the links among dementia, altered mobility, and balance were clear, other studies around the same time began finding increased evidence for links between the bone and the brain through the effects of psychotropic drug treatments. Investigators found an increased risk of hip fracture in patients taking several classes of long-acting psychotropic drugs from 1980 through 1982, even after controlling for confounding dementias.[22] Clinical observations of accelerated fracture healing in patients with head injuries in the late 80s were one of the most surprising pieces of evidence linking bone and brain communication, but these studies were small and lacked molecular or cellular evidence.[23,24] By the mid-1990s, new classes of growth factors (e.g., bone morphogenetic proteins [BMPs] and the transforming growth factor-β [TGF-β] superfamily) with known roles in skeletal development were discovered. Surprisingly these factors were also discovered in brain tissue and were linked to important growth and regulatory mechanisms in neural tissue, laying the foundations of molecular connections between the nervous system and the skeleton.[25,26] Concurrently, studies focusing on the fracture risk in patients diagnosed with AD and PD found an increased risk of fracture in their cohorts but attributed these to increased fall risk and not disuse osteoporosis,[27–29] although follow-up studies in the 2000s concluded that concomitant dementia and falls did not entirely explain fracture risk in these patients.[30,31] In 2004, a study of falls in elderly Canadians ≥65 years of age (N=1,513) between 1994 and 1995 was the first to find an independent relation between AD and hip fractures.[32] Most significantly, Weller and Schatzker [32] found that the odds ratio relating AD and hip fractures were stable and only slightly changed after accounting for falling and osteoporosis, implicating factors other than just a loss of physical mobility and stability with hip fracture in patients with AD and ADRD. Later studies confirmed that individuals who experienced fractures had a higher risk of later developing dementia.[33] A meta-analysis in 2012 of nine separate clinical studies found a greater risk of hip fracture and lower bone mineral density (BMD) in patients with AD than healthy controls, reinforcing the clinical evidence that factors beyond falls, impaired mobility, and loss of balance were behind the association of bone fracture and dementia.[34] By 2019, it was noted that patients receiving treatment for low bone mass or osteoporosis more often also presented with accelerated loss of brain volume or parenchymal atrophy in magnetic resonance imaging scans, even when there was no history of dementia.[35] With mounting clinical evidence, the need to study the molecular basis behind the newly coined ‘bone-brain’ axis was becoming clear.
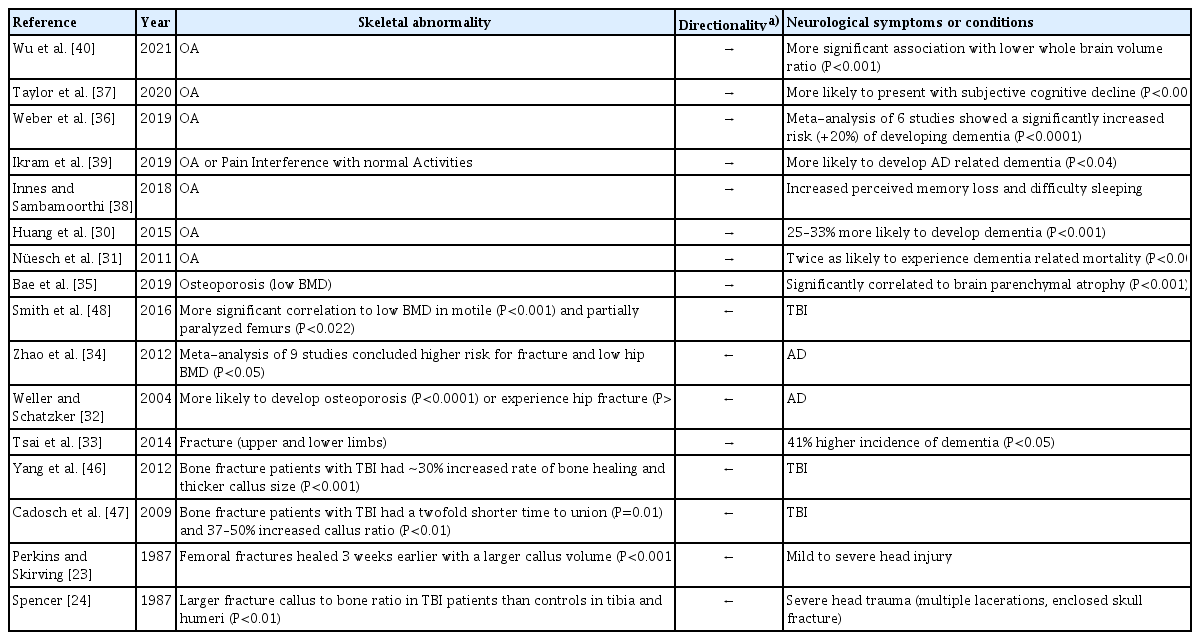
Clinical observations/evidence for bone-brain disease relationships
Throughout the 2000s other skeletal diseases, such as OA - a disease of the whole joint in which multiple tissues are affected by and contribute to the degeneration of articular cartilage and subchondral bone - also began being associated with cognitive decline. By the 2010s, clinical investigations began linking dementia to patients with OA, with some marking an increased risk of developing dementia when diagnosed with OA or even observing an increased risk of dementia-associated deaths in patients with arthritis.[30,31] By 2019, a meta-analysis of 6 independent studies, including 388,252 individuals, after controlling for age and gender, concluded that there was a roughly 20% increased risk (P<0.0001) for developing dementia when a patient had or previously presented with OA.[36] Conversely, the Centers for Disease Control and Prevention reported in 2020 that, in the USA, 60.3% of adults aged 45 to 65 years and 63.9% of adults aged 65 years or older with arthritis also experience subjective cognitive decline, one of the earliest noticeable symptoms of dementia, as well as memory and sleep loss.[37,38] Even perceived pain in the joints without a diagnosis of OA was linked to a higher likelihood of developing AD or ADRD as reported in 2019.[39] Recently, in a neuroimaging study, OA, even in non-demented patients, was associated with a greater loss in gray matter volume of the whole brain than in other non-demented elderly individuals without OA.[40] The hypothesized overlap in pathological cytokine profiles between inflammatory conditions, such as OA and AD, extends the bone-brain axis to include cartilaginous joints.
Beyond AD, several other conditions have been linked to skeletal abnormalities. In a large cohort study of Korean patients with osteoporosis, patients presented with a 1.49-fold higher occurrence of PD than patients without osteoporosis.[41] In another study, osteoporosis and osteopenia were common findings in patients with PD, affecting up to 91% of women and 61% of men.[42] Huntington’s disease (HD), a genetically linked neurodegenerative disorder, was also associated with lower bone mass in selected clinical studies even after correcting for confounding factors, such as past medical history, drug history, diet, and levels of physical activity.[43,44] Skeletal conditions in genetic models of neurodegeneration, such as HD, signify the existence of cross-talk between the bone and brain beyond solely inflammatory mechanisms.
One of the most compelling pieces of evidence for complex signaling mechanisms between the nervous system and the skeleton is the paradoxical occurrence of skeletal pathologies alongside traumatic brain injury (TBI). In some clinical observational studies, increased rates of fracture healing and fracture callus size were seen in individuals who experienced TBI in conjunction with a fracture.[45–47] Paradoxically, other patients who experienced TBI alone without a fracture were more likely to suffer from bone mass loss or develop osteoporosis after or in conjunction with injury.[48] The contradictory nature of the findings in bone and brain injury, and the rarity of observing these co-incident injury events in humans, prompted investigators to recapitulate these phenomena in murine models. After combining an osteotomy, surgically induced bone fracture, with a controlled cortical impact to induce TBI in mice, the combined-trauma groups showed greater bone volume, higher mineral density, and a higher rate of fracture-gap-bridging than the control group which featured the fracture alone without TBI. Mice in the combined trauma groups even demonstrated increased torsional strength in the femur around the healed fracture after 4 weeks, indicative of accelerated healing that normally is achieved after 5 to 6 weeks in mice.[49] In contrasting studies, mice exposed to repetitive mild TBI alone presented with reduced bone volume and density and lowered peak torsional strengths in the vertebrae and femur.[50] The many different types of skeletal comorbidities with several independent neurodegenerative conditions and their complex relations to one another, documented in clinical studies and observations establish the existence of the bone-brain axis. Understanding the role that the skeletal system and the brain play in neurodegenerative and skeletal diseases highlight the need for a greater understanding of the signaling mechanisms within this system.
PHYSICAL, MOLECULAR, AND FUNCTIONAL SIMILARITIES BETWEEN BONE AND BRAIN CELLS
As clinical evidence for the bone-brain axis began emerging in literature near the end of the 20th century, investigators began looking for causal relationships behind these relationships on the cellular and molecular levels. In addition to the well-known and striking physical similarities between cell types specialized to discrete functions in these 2 organ systems, cellular and molecular evidence adds important insight into the interplay of bone and brain cells as well as the direct innervation of bone tissue, the migration of bone cells to the brain, and shared utilization of specific signaling pathways and signaling factors in both organ systems.
1. Osteoclasts and microglia
Osteoclasts and microglia are tissue-resident and -specific cells of monocyte/macrophage lineage in skeletal and neural systems, respectively, that physically regulate their resident microenvironments (Fig. 1A). Osteoclasts are large multinucleated macrophage-like cells that degrade the bone matrix with hydrogen ions and catalytic enzymes as the initial player in bone remodeling. Microglia are specialized brain-resident macrophages responsible for the selective elimination of inappropriate synaptic connections during neural development and remodeling of synaptic circuits in adulthood.[51,52] Both cell types trace their origins to the same initial site of extraembryonic hematopoiesis in the yolk sac (YS) during embryogenesis.[53] These YS-derived cells become hematopoietic stem cells (HSCs) that, in turn, produce macrophages, erythrocytes, and lymphocytes, and give rise to microglia in the central nervous system (CNS).[54,55] Additionally, YS-derived HSCs generate bone marrow, the main source of hematopoiesis in adult life and the main site of most osteoclast precursors.[56,57] The shared functional roles of osteoclasts and microglia in tissue remodeling implicate them in pathological changes to their resident tissues, playing causal roles in degenerative diseases, osteoporosis, and AD, respectively.
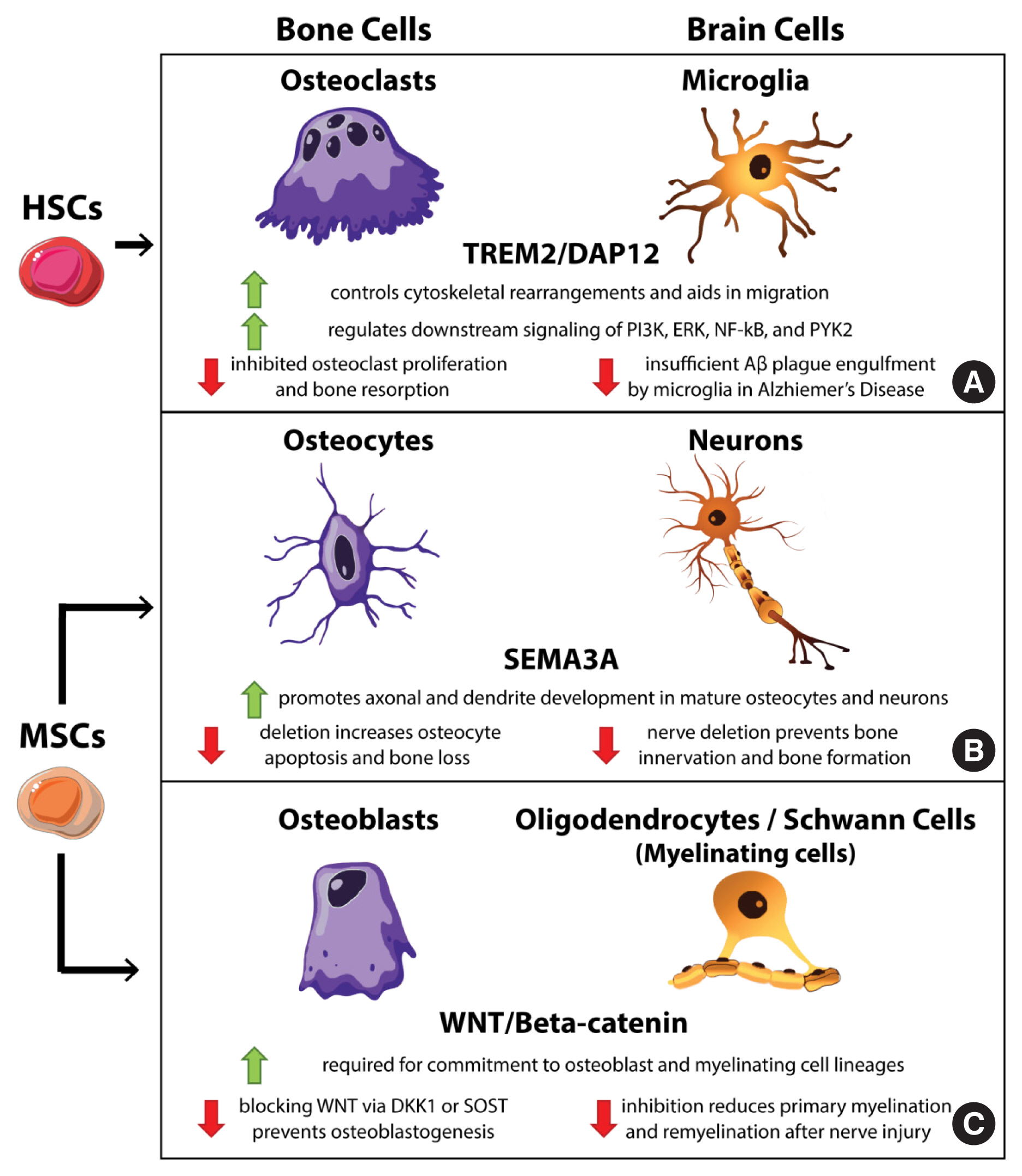
Cellular analogs in bone and the brain. Specialized cells within the bone and the brain have phenotypic and functional analogs in the reciprocal organ system that share certain signaling mechanisms. Osteoclast and microglia (A) are resident macrophages of bone and brain, respectively, that share hematopoietic stem cells (HSCs) origins and remove materials from their resident tissues to maintain proper tissue function. Both are affected by the triggering receptor expressed on myeloid cells 2 protein (TREM2)/DNAX-activating protein of 12 kDa (DAP12) signaling pathway. Osteocytes and neurons (B) share a dendritic and axonal phenotype and also serve as sensory cells throughout the body and the skeleton. Both cell types utilize semaphorin 3A (SEMA3A) to direct axonal projections and are derived from mesenchymal stem cells (MSCs). Osteoblast and myelinating cells (oligodendrocytes and Schwann cells) (C) produce materials that support the functions of their resident tissues and require Wingless/Integrated-1 (Wnt) signaling for proper differentiation from their MSC origins. P13K, phosphoinositide 3-kinases; ERK, extracellular signal-regulated kinases; NF-κB, nuclear factor-κB; PYK2, protein tyrosine kinase 2; DKK1, dickkopf Wnt signaling pathway inhibitor 1; SOST, sclerostin.
Beyond origin and functional roles, osteoclasts and microglia utilize several of the same signaling mechanisms. Both rely on similar growth factors and signaling molecules for proliferation and migration, including colony-stimulating factor-1, C-C motif chemokine receptor 5, and protein tyrosine kinase 2 (PYK2). Mutations or deletions within these genes result in pathologies including osteopetrosis, osteoporosis, progressive cognitive decline, motor disturbances, seizures, and the inability to regulate tau pathologies in AD.[53,58–64] Another important regulatory mechanism shared by these 2 macrophages is the triggering receptor expressed on myeloid cells 2 protein (TREM2)/DNAX-activating protein of 12 kDa (DAP12) signaling pathway (Fig. 1A). Blocking TREM2/DAP12 signaling in osteoclasts leads to lower bone resorption, while stimulation enhanced osteoclast migration.[65–67] In microglia, TREM2/DAP12 signaling mediates phagocytosis.[68] Dysfunction within this complex leads to insufficient amyloid β (Aβ) plaque engulfment.[69,70] TREM2/DAP12 has also been utilized in genetic models of AD and PD to fight disease progression and neural inflammation.[71,72] Signaling through the TREM2/DAP12 complex activates several other shared pathways in osteoclasts and microglia, including Src tyrosine kinase, phosphatidylinositol 3-kinase, extracellular signal-regulated protein kinase (ERK), nuclear factor-κB (NF-κB) and PYK2.[53] For a more complete discussion of these signaling factors and their overlap and involvement in disease and pathologic conditions linked to these cell types, see Lee et al.[53] Important to the discussion of the bone-brain axis of signaling, communication through the TREM2/DAP12 pathway activates ɣ-secretase, which generates a soluble form of TREM2 (sTREM2) that can cross the blood-brain barrier in conditions of neural degeneration where the integrity of the BBB is compromised and create a peripheral axis of bone to brain signaling.[73] The existence of this self-propagating signal suggests a potential role of peripheral, osteoclast-generated sTREM2 in TREM2 biology and microglia function in the brain, providing a direct route of signaling within the bone-brain axis.
2. Osteocytes and neurons
Since the late 90s investigators have asked if bone cell networks mimic neuronal circuits in form and function.[74, 75] Visually, the complex networked appearance of the osteocyte lacunar-canalicular network (LCN) calls to mind the bundles of nerves and synaptic connections within the brain and the nervous system (Fig. 2). While qualitative efforts to compare the size of these 2 cellular networks are based on estimates, the magnitudes of the osteocyte LCN and neuronal networks are thought to be similar. The end-to-end length of all myelinated nerve fibers in the brain is estimated to be around 176,000 km long in a mature adult human male,[76] which is almost exactly the estimated end-to-end length of the LCN calculated by Buenzli and Sims [77] at 175,000 km. When compared to the vasculature, the next longest cellular network in the body, which is estimated to be around 100,000 km end-to-end,[78] nerve and osteocyte networks place within the longest and largest cellular networks within the body. Other size-related parameters emphasize the sheer size and similarities of the 2 networks. There are 78 to 97 billion individual neurons within the brain with approximately 100 trillion cell-cell connections (synaptic or otherwise) that cover a surface area of around 200 m2.[79–83] The osteocyte LCN appears at a slightly smaller but comparable magnitude with an estimated 33 to 50 billion cells with a total of 23 to 24 trillion cellular gap junction connections that covers a slightly larger surface area of 215 m2.[77] The comparable network-like geometry of connections within nerves and the osteocyte LCN has also lent them to similar tools for quantification and analysis. Mathematic models relying on graph theory, termed connectomics network analysis, that were developed to map the intricate networks within and between brain cells in homeostasis and disease [84–86] have been effectively applied to similar studies of the osteocyte network to map complex geometries and network topology.[87–90] Nerve and bone networks share many physical similarities, including size and geometry.

Comparison of the osteocyte lacunar-canalicular network (LCN) and neural networks. Osteocytes (A) and neurons (B) share a similar cellular morphology of a central larger cell body with axonal projections. These projections extend and connect to other cells to create the LCN for osteocytes or the greater nervous system. These cellular networks each extend for approximately >175,000 km within an adult human and form the longest networks in the body and are each responsible for sensing and responding to external cues.
The relationship between nerves and osteocytes extends past form to function. The roles of nerves to send and receive signals throughout the body are well understood and so is the ability of certain specialized nerve cells to physically sense different stimuli, such as pressure, light, and temperature.[91] Internal cortical bone lacks a large amount of internal innervation yet has the unique ability to sense and respond to mechanical stress independently of sensory nerves.[92] Osteocytes within the LCN sense the motion of pericellular fluid between themselves and the mineralized walls of the LCN as drag, shear forces, or stretch and respond by secreting signaling factors that increase osteoblastic bone deposition and inhibit osteoclast bone resorption.[93,94] Cessation or long-term absence of these mechanical signals through sedentary lifestyles, limb immobilization, or microgravity in spaceflight results in the opposite effect with osteocytes producing factors that increase osteoclastic bone resorption and bone loss. Because of their release into circulation, many of these secreted factors have distal effects on other cell types, including potential roles in the nervous system, discussed later and in more detail by Gerosa and Lombardi [95]. The role of the osteocyte network as a mechanosensory organ often grants it the title of ‘brain of the bone’.
In addition to morphological and some functional similarities between osteocytes and nerves, several growth factors act on both cell types to direct growth and development. Dendrite formation and guidance are important biological functions necessary in the development of the nervous system and the osteocyte network to establish the proper cell-to-cell connections upon which both sets of cells rely. One class of these axon guidance factors are the semaphorins, which have a variety of roles in neuronal patterning.[96] Specifically, semaphorin 3A (SEMA3A) (Fig. 1B), released by sensory nerves, is a diffusible axonal factor that acts as a chemorepellent during development but as a chemoattractant in adult nerve cells.[97] Nerve-specific deletion of this factor resulted in osteopenia in mice due to lost innervation of developing bones, but there was no effect in osteoblast-specific deletion demonstrating the role of nerve-derived SEMA3A and the importance of bone innervation in growth.[98] Interestingly, SEMA3A may have additional roles in bone independently of nerve cells through the development and maintenance of osteocyte networks. Treatment of MLOY4 osteocyte like cells in vitro with soluble SEMA3A promoted dendrite development,[99] whereas direct deletion of SEMA3A in mature osteocytes resulted in increased osteocyte apoptosis and bone loss.[100] Another factor that promotes osteocyte dendrite production and growth in vivo is the transcription factor Sp7, also known as osterix, which enhances the expression of genes involved in dendrite elongation and axon guidance. Sp7 in osteocytes is a transcription factor that regulates expression of the SEMA3A gene and many other factors involved in axon guidance and dendrite development. Accordingly, deletion of Sp7 in osteocytes via DMP1-Cre methods suppresses expression of several genes including SEMA3A and other semaphorins.[101] Osteocyte-specific deletion of Sp7 also resulted in osteopenia, similar to results observed when SEMA3A was deleted specifically in sensory nerves.[98,101] While the precise mechanisms regulating bone innervation and osteocyte network development remain to be explored, the utilization of SEMA3A by both nerves and osteocytes to generate a shared cellular feature, dendritic projections, independently of one another highlight molecular similarity and potential for crosstalk in both sensory nerves and bone-embedded osteocytes.
The expanded role of the skeleton as an endocrine organ stems from the utilization of typical neuronal factors by osteocytes. Neuropeptide Y (NPY) is a 36–amino acid polypeptide and regulator of energy homeostasis that is primarily expressed throughout the CNS and peripheral nervous systems (PNS) that has roles in managing anxiety and stress, depression, energy homeostasis, and memory.[102,103] Through its broad class of 5 NPY receptors (NPYRs) that are specifically expressed among different cell types, NPY is able to act in non-neuronal tissues to regulate blood pressure, heart rate, digestion, body metabolism, and immune functions.[104] NPY modulates bone and body mass in an endocrine fashion through ghrelin singling in the hypothalamus by controlling food intake.[105–107] Peripheral NPY may act directly on bone marrow mesenchymal stem/stromal cells (BMSCs)—osteoblastic precursors—to control osteogenesis and adipogenesis.[108] NPY directly affects bone mass by signaling through NPYRY1 (Y1) expressed on osteoblasts. Over-expression of NPY to increase Y1 signaling reduced bone mass. Conversely, inhibiting NPY signaling through deletion of Y1 in osteoblasts enhanced bone formation.[109,110] NPY is also produced directly in the skeleton by osteocytes and acts as an inhibitory signal to reduce bone formation by osteoblasts.[111] Given their proximity to the vasculature and secretory capacity, osteocytes may use NYP to signal back to the NPYRY2 (Y2) expressing NPY neurons in the hypothalamus where the BBB is the weakest [112,113] as a bone-to-brain mechanism of signaling to modulate systemic metabolism and bone and body mass. While the bi-directionality of NPY signaling between osteocytes and brain cells remains to be fully explored, the utilization of this pathway in both neurons and osteocytes to mediate bone and body mass emphasizes more systemic functional similarities between these 2 cell types.
3. Osteoblasts, Wnt signaling, and the brain
While osteoclasts and osteocytes have more direct analogs within the nervous system, a direct counterpart for osteoblasts, the bone-forming cell, is less obvious. Osteoblasts are derived from MSCs that give rise to mesodermal-lineage cells, including cartilage and adipocytes, as well as bone, but also into other lineages of ectodermal and endodermal cells. Differentiation towards mature osteoblasts occurs through the successive activation of key regulators including Sox9, Runx2 - a transcription factor essential for osteoblastogenesis, followed by Sp7/osterix, and ultimately commitment to the osteoblast lineage.[114,115] Many other key regulatory pathways take part in osteoblastogenesis including Hedgehog and BMP signaling. Given the necessity of Wnt/β-catenin signaling for the activation of the Runx2 promoter, Wnt activation is often thought to be one of the main determinants of osteoblastic cell fate, especially since its activation also inhibits the expression of adipogenic transcription factors and blocks preadipocyte differentiation.[115,116] Given the mesenchymal background and utilization of BMP and Wnt signaling mechanisms, mechanisms utilized by almost all cell types in differentiation, the selection of a unique analog within the brain for osteoblasts is difficult. However, there are biological and functional similarities to consider in the comparison of osteoblasts and neuronal cells in the context of bone – brain signaling.
Functionally, mature osteoblasts are responsible for the deposition of the initial bone osteoid that is remodeled into mature bone and also gives rise to osteocytes from osteoblasts that become embedded in their matrix during the bone-forming process. Unlike osteocytes and osteoclasts, functional comparisons for osteoblasts to the remaining brain cell types, astrocytes and oligodendrocytes or other myelinating cells, is more strained. For example, oligodendrocytes and Schwann cells, the glia cell types responsible for myelinating nerve fibers in the CNS and PNS, respectively, could be used as comparative functional analogs for osteoblasts in the brain (Fig. 1C). These 2 cell types are functionally similar in that both cell types provide structural roles for their ‘home’ tissues: osteoblasts deposit the bone matrix, and myelinating glia cells enwrap and support nerve cell axons. Osteoblasts and glial cells are also both responsible for repair and reconstruction of their microenvironments after injury. Osteoblasts help to form the mineralized fracture callus, and glial cells direct and support nerve repair after.[117] In medaka, a Japanese rice fish, it was demonstrated that myelinating glial cells colocalize with osterix expressing osteoblasts during bone fracture healing. Inhibition of this synergy via rapamycin treatment prevents clusters of these cells from forming, and subsequently a non-union occurs.[118] While the exact signaling mechanism and broad effects of rapamycin treatment in this study [118] remain to be elucidated, the functional cooperation of myelinating glial cells and osteoblasts in fracture healing raises additional questions about their relevance in the bone-brain axis. Functionally, osteoblasts and glial cells share important structural and ECM regulatory roles in their respective tissues.
While the functional comparison between osteoblasts and myelinating glial cells is intriguing, it requires more experimental evidence; however, a more important discussion revolves around shared cellular and molecular regulators of osteoblasts and neural cells. For example, in investigations involving zebrafish, Schwann cell precursors were seen to migrate away from peripheral nerves during embryonic development to become general MSCs within the neural crest where they contribute to the generation of cartilage and bone in the craniofacial skeleton, thoracic spine, and ribs.[119] This type of transdifferentiation and shared lineage raises questions about the molecular regulators of osteoblasts and neural cells. Typically, glial cells, such as oligodendrocytes and Schwann cells, both rely on Wnt/β-catenin signaling for appropriate differentiation.[120,121] In addition, blocking Wnt signaling in Schwann and oligodendrocytes cells (1) prevents myelin gene expression; (2) represses overall myelinogenesis; and (3) inhibits remyelination after nerve injury.[122,123] While Wnt signaling is an ancient and evolutionarily conserved pathway that regulates many aspects of cell fate and cell determination in many cell types, it is of particular interest in the brain for its relation to both homeostasis and neurodegeneratsuspect in the bone-brain axis and the clinicalion. While some specific cell types, such as the myelinating cells, require Wnt signaling for appropriate cell-fate decisions other neural cells are also susceptible to signaling molecules that traditionally target osteoblastic Wnt signaling. Thus, Wnt signaling in general has become an important topic in bone-brain axis research.
In canonical Wnt signaling, Wnt1 class ligands, such as Wnt1 and Wnt3a, bind a complex of Wnt receptors, including frizzled, lipoprotein related peptide (LRP)5, and LRP6. Binding of this complex phosphorylates, and thus inhibits, the intracellular mediator glycogen synthase kinase-3β (GSK-3β). In its uninhibited state, GSK-3β phosphorylates cytoplasmic β-catenin, a translatable transcription factor, to target it for ubiquitination and degradation. Thus, activation of Wnt receptors and the inactivation of GSK-3β induces the accumulation and translocation of collected β-catenin into the nucleus to induce transcription of target genes.[124–126] In skeletal precursors, β-catenin enhances the differentiation of precursors into osteoblasts. Several molecules inhibit this signaling pathway by blocking interaction with the Wnt receptors. In bone, DKK1 and osteocyte-produced SOST bind the LRP5/6 complex and inhibit Wnt signaling in osteoblasts, limiting osteoblastogenesis and favoring bone resorption.[127] In the brain, Wnt signaling is a powerful repressor of neuroinflammation in several ways. Signaling through the Wnt receptor LRP5 produces anti-inflammatory macrophage phenotypes in microglial cells.[128] Wnt signaling and β-catenin translocation in microglial cells physically prevents the expression of inflammatory markers from the NF-κB signaling pathway and directly induce expression of PPARγ, a well-known anti-inflammatory agent that itself also antagonizes NF-κB-mediated transcription of downstream genes.[129,130] Thus, Wnt signaling in the brain exerts its anti-inflammatory activity in multiple ways, and inhibition may produce dangerous inflammatory conditions. Indeed, disruptions to Wnt signaling in the brain have been linked to synaptic disassembly of neurons and the breakdown of the BBB, critical malformations in neurodegenerative diseases.[131,132] Thus, disruption or inhibition of Wnt signaling has been implicated across multiple cell types in a panoply of neurodegenerative diseases, including AD, PD, HD, and multiple sclerosis, as well as in complications related to healing after ischemic stroke.[133] Given the importance of Wnt signaling for protecting against neuroinflammation and neurodegenerative disorders and the large source of secreted Wnt -inhibitors from the bone, Wnt signaling has become a prime suspect in the bone-brain axis and the clinical relationships between comorbidities of osteoporosis and brain disorders. Dkk1 is increased in the brains of AD patients and active in the dispersal of synaptic components in AD [134] and, while mechanisms of transfer of SOST to the brain may be accomplished in neurodegenerative disorders where the BBB is degraded, direct injection of SOST into mouse brains produced anxiety-like behaviors and reduced dendritic length and branching in pyramidal neurons of the hippocampus.[135] Further investigation on the pathological role of bone-derived Wnt inhibitors on neurodegenerative diseases is warranted, but the shared utilization and negative impact of disrupted Wnt signaling in the skeleton and the brain provide another molecular link within the bone-brain axis.
In addition to bone-derived circulating factors, some bone-derived cells travel to the brain, establish residence, and impact neuronal health. The bone marrow is a large source of mesenchymal stem cells in the body that gives rise to a range of cell types, including mature bone and brain cells. Some BMSCs have a unique ability to migrate to the peripheral blood and then make their way to the brain, where they cross the BBB when it is damaged by stroke or disease (Fig. 3).[136] These BMSCs include precursors for mesenchymal stem cells that can partially differentiate into nerves, bone marrow-derived microglia-like cells (BMDMLs) and general bone marrow macrophages (BMMs) that aid in clearance within the brain.[137] The use of BMSCs and BMDMLs in AD mice improves learning and memory, limits further Aβ plaque formation, and promotes rapid clearance of Aβ, which in turn improved cognitive dysfunction.[138,139] Transplanted BMSCs via intranasal delivery in mice and rats are beneficial in PD in repairing neuronal tissue after ischemic stroke and in reducing neuropathic pain by combating chronic inflammation.[137] Interestingly, in situations of chronic stress, BMDMLs were seen to migrate to the paraventricular nucleus (PVN) in the hypothalamus, express the proinflammatory factor interleukin-1β and signal to glutamate receptors in PVN neurons inducing depression and anxiety-like behaviors in mice. [140] Whatever their role, the surprising ability of bone-derived cells to migrate to the brain, establish residence, and affect local and systemic conditions demonstrates a direct cellular portion of the bone-brain axis that offers many avenues for the treatment of neurodegenerative and psychologic disorders.
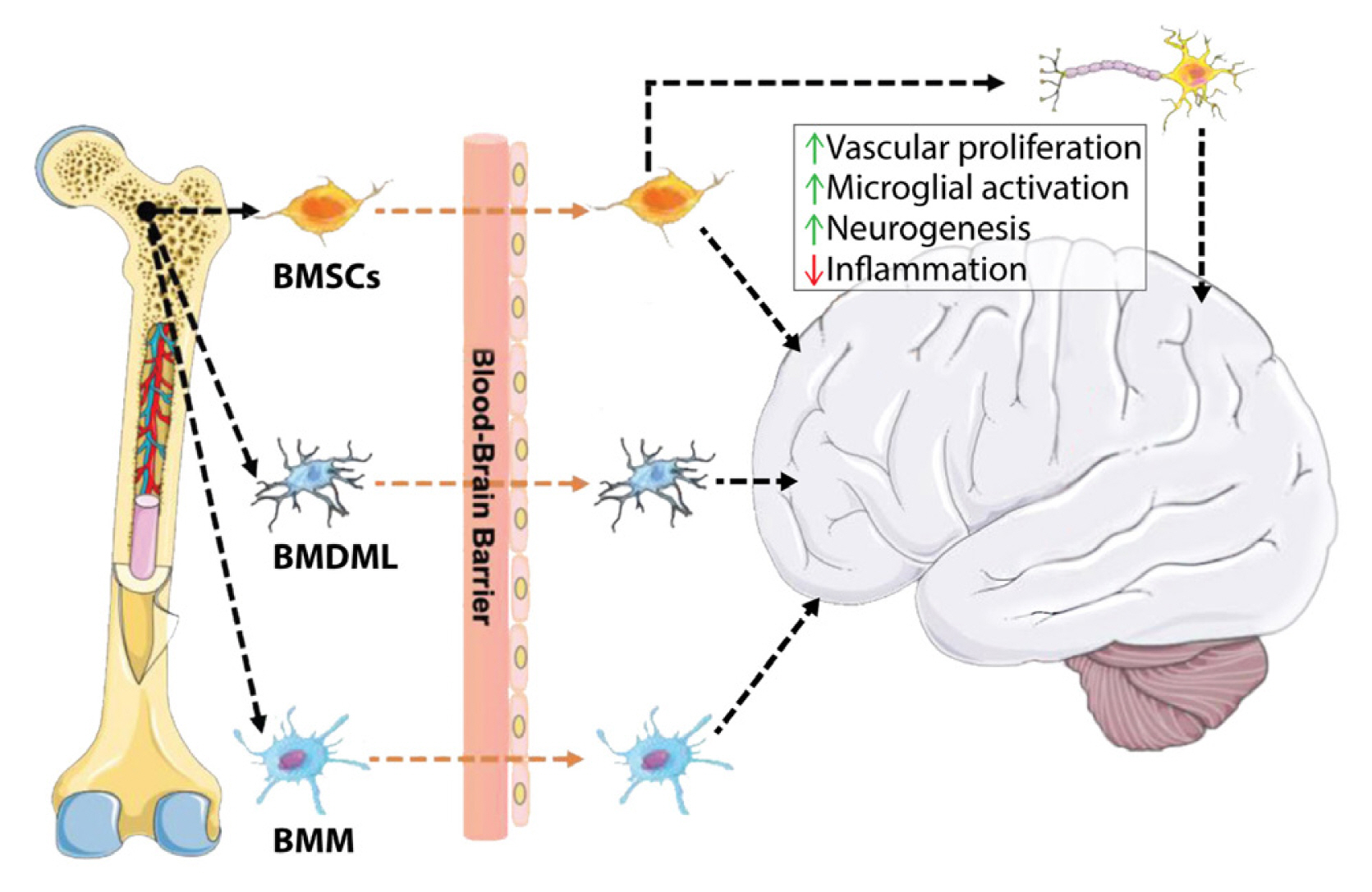
Routes of direct cellular migration from the bone to the brain. The cellular mechanism of central regulation by bone-derived cells. BMSCs, bone marrow mesenchymal stem/stromal cells; BMDML, bone marrow-derived microglia-like cells; BMM, bone marrow macrophages. [Modified from “Inspiration for the prevention and treatment of neuropsychiatric disorders: New insight from the bone-brain-axis.”, by Zhao Y, et al., 2021, Brain Res Bull, 177, pp. 263–272. Copyright 2021 by the Elsevier Science. Modified with permission].
The utilization of many similar signaling pathways, including the Wnt pathway, and their similar stem cell sources link bone and brain beyond simple comorbidity in disease. Several cell types from each tissue mirror each other in function and susceptibility to similar molecular regulators. Direct cellular innervation, connection, or migration may influence the conditions of the reciprocal tissues. Thus, strategies to intervene in bone or brain have the potential to result in commensurate effects in the opposite tissue, prompting further research into the bone-brain axis.
BRAIN TO BONE AND BACK AGAIN: BI-DIRECTIONAL AVENUES FOR COMMUNICATION
The existence of the bone-brain axis has been recognized for decades. The first review on the topic appeared in 2004 and discussed the role of several neural ligands in clinical conditions, such as heterotopic bone formation, musculoskeletal pain, and obesity and bone mass, among others.[141] Since then, knowledge of neuro-skeletal crosstalk has expanded beyond traditional unidirectional brain-to-periphery communication utilizing neurotransmitters (NTs) to include bi-directional communication through several different classes of signaling molecules.[142] Traditionally, distal signaling from the body to the nervous system is considered highly exclusive due to the existence of the BBB that prevents most small molecules from entering the CNS. In recent years, certain privileged molecules from the skeleton have been suggested to enter the CNS and bypass the BBB by direct transport.[12] Here we briefly review the several classes of signaling molecules that compose the bone-brain axis (Fig. 4).

Bidirectional molecular communication between bone and the brain. The bone-brain axis involves bidirectional communication between the brain and bone that utilizes several classes of signaling factors. Neural tissues produced several neurokines, including neurotransmitters, neuropeptides, and neurohormones that signal down from the brain to bone cells. Several bone-derived osteokines also signal “upward” from the periphery to the brain through the circulation and bypass the blood brain barrier in homeostasis and disease to affect neural cell behavior and systemic metabolism. NE, norepinephrine; CgRP, calcitonin gene-related peptide; SP, substance P; SEMA3A, semaphorin 3A; FSH, follicle-stimulating hormone; TSH, thyroid stimulating hormone; ACTH, adrenocorticotropic hormone; GH, growth hormone; AVP/ADH, arginine vasopressin/antidiuretic hormone; OT, oxytocin; NPY, neuropeptide Y; AgRP, agouti-related peptide; CART, cocaine- and amphetamine-regulated transcript; VIP, vasoactive intestinal peptide; OCN, osteocalcin; LCN2, lipocalin2; SOST, sclerostin; DKK1, dickkopf Wnt signaling pathway inhibitor 1; FGF23, fibroblast growth factor 23; OPN, osteopontin; RANKL, receptor activator of nuclear factor-κB ligand; BMP, bone morphogenetic protein; IGF-1, insulin-like growth factor 1.
1. NTs
The first major class of signaling molecules that compose the bone-brain axis are the NTs (Fig. 4) that act directly on bone cells and in an endocrine-like fashion to regulate bone cell metabolism. While it has been long recognized that portions of the bone, including the periosteum, bone marrow, and vascular canals are innervated directly by nerves, [16,17] their precise signaling mechanisms and roles in bone are only recently being understood. Several NTs, more extensively reviewed by Gerosa and Lombardi [95], are delivered directly to the bone by a collection of parasympathetic, sympathetic, and sensory nerves. These include the muscarinic and nicotinic signaling molecules acetylcholine and norepinephrine (NE) that act via α- and β-adrenergic receptors, as well as several other specialized signaling molecules the semaphorins, calcitonin, and other small peptides, such as substance P (SP). Primarily, stimulation of bone cells by parasympathetic acetylcholine signaling promotes bone formation while signaling via NE from the sympathetic nervous system promotes bone resorption.[143, 144] In addition, several of these signaling factors, especially those from sensory nerves, have roles in bone formation and bone healing after fracture where denervation studies showed delayed healing and smaller callus size.[145] The effects of other NTs can operate in complex fashions, such as brain-derived serotonin that indirectly acts through leptin to increase bone formation, while peripherally produced serotonin directly inhibits osteoblastogenesis and bone formation.[146] The roles of another endocrine-like NT, glutamate, have been hypothesized to be involved in the mechanosensitive regulation of bone mass.[147] The role of dopamine signaling in the healthy skeleton has yet to be fully elucidated, though treatment with the atypical antipsychotic drug Risperidone, an inhibitor of dopaminergic signaling, had detrimental effects on BMD in patients.[148] Additionally, dopamine may directly act to increase bone mass by interacting through the ERK1/2 pathway.[149] NTs are a broad class of small molecules that provide a direct route of communication from nerve cells to bone.
2. NPs
Another class of molecules that mediate signals from the brain to the bone are the NPs (Fig. 4). NPs are small chains of amino acids generated from larger precursor prepropeptides and are primarily synthesized by neurons; however, some forms of NPs are produced by other peripheral cell types, including bone-resident osteocytes as discussed.[111] NPs are often packaged alongside NTs in separate dense core vesicles and are released from axons simultaneously with other signaling molecules to act directly in the brain and on distal innervated tissues. Like other neurokines, NPs have a broad range of effects on the skeleton by targeting several cell types within the CNS and the rest of the body. NPs may exert their influence either directly through their respective receptors on distal cells or through an endocrine like-fashion by activating different brain regions. This is exemplified by melanocortin for which mutations in its receptor in the hypothalamus result in genetic obesity, which negatively impacts bone mass, while direct signaling to its receptor on osteoblasts has osteoinductive effects. [150,151] Other endocrine-like NP mechanisms include neuromedin U (NMU) where abolished NMU signaling prevented leptin- or sympathetic nervous system–mediated inhibition of bone formation with simultaneous altered expression of molecular clock genes in bone cells, demonstrating a larger endocrine regulation of bone cells via neural circuits.[152] Other NPs, such as cocaine amphetamine-regulated transcript, vasoactive intestinal peptide, calcitonin gene-related peptide, and SP affect bone health by intersecting with other well-known bone regulatory mechanisms, such as receptor activator of NF-κB (RANK) ligand (RANKL), and osteoprotegerin signaling that balances osteoclast action (reviewed in more detail elsewhere [11,153–155]). Targeted dysfunction of other neural circuits utilizing NPs in the arcuate nucleus of the hypothalamus, including neurons expressing agouti-related peptide and Kisspeptin, also generate bone-specific phenotypes. Disruption of agouti-related peptide neurons resulted in lost bone mass while lost action in Kisspeptin circuits involving estrogen signaling dramatically increased bone mass.[156–158] While the downstream signaling mechanisms ultimately affecting bone mass remain to be elucidated in these cases, they represent powerful NP signaling mechanisms that regulate bone health. Similar to more traditional NTs, NPs provide another molecular route of brain-to-bone signaling.
3. Neurohormones (NHs)
A third class of brain-derived signaling molecules is the NHs that are released by the hypothalamus in the CNS directly into circulation to influence skeletal health peripherally (Fig. 4). Receptors for these secreted small molecules exist on either or both osteoclasts or osteoblasts and exert their effects to modulate bone resorption and formation. Some NHs, like thyroid-stimulating hormone, are highly context-dependent as it has been shown to inhibit both osteoclastogenesis and osteoblastogenesis but also promote osteoblast proliferation through non-canonical Wnt signaling.[159] Other NHs preferentially activate one side of the bone remodeling unit, either formation or resorption, such as follicle-stimulating hormone that promotes resorption by having its receptors only expressed by osteoclasts and their precursors. To add further layers of complexity, some NHs may act indirectly on bone cells in addition to their direct binding of bone cells by stimulating the release of other signaling molecules, as is the case for growth hormone (GH) that predominantly exerts its function through insulin-like growth factors (IGFs).[160] The ability of NHs to influence the skeleton via circulation reveals a systemic brain-to-bone signaling mechanism within the bone-brain axis.
OSTEOKINES
Osteokines comprise the reciprocal axis of peripheral signaling from bone to the brain. These are bone-cell-derived signaling molecules, each with a specified function within the bone compartment, many of which are produced and released by osteocytes as a part of their regulatory role in bone metabolism (Fig. 4). Osteocytes release these factors via extracellular vesicles (EVs) for communication to bone cells, but these EVs may also reach circulation through the osteocytes’ close ties to the vasculature. Fluorescently labelled osteocyte EVs even cross the BBB of mice on relatively short timescales (6 hr) and have enhanced uptake in neurodegenerative models where the BBB may be degraded and more permeable.[161] The serum levels of several bone-derived signaling factors have been measured as elevated in individuals with osteopenia and osteoporosis, and several other bone-specific markers (e.g., osteocalcin [OCN], osteopontin [OPN], SOST, DKK1, BMPs) are also associated with neurodegenerative diseases, such as AD and PD.[13,162–165] Given the prevalence of these bone-derived signaling molecules in neurodegenerative disease and the ability for bone cells to transmit factors from bone to circulation and ultimately bypass the BBB, osteokines represent the signaling from the bone periphery to the brain portion of the bone-brain axis. We discuss several osteokines in detail below.
1. OCN
OCN, also known as bone γ-carboxyglutamic acid protein, is produced by osteoblasts and aids in the association of bone mineral to collagen to improve bone strength and quality. Due to its susceptibility to enzymatic degradation, several forms of OCN may be found in circulation and is thus a circulating marker of bone formation.[166] Systemically, non-carboxylated OCN is thought to play roles in insulin secretion, testosterone synthesis, and muscle formation. Recently, circulating forms of OCN were seen to cross the BBB in the brainstem, midbrain, and hippocampus and impact the synthesis of monoamine NTs, including serotonin, dopamine, and noradrenaline, and inhibit the synthesis of γ-aminobutyric acid (GABA).[163,167] In OCN null mice, peripherally administered non-carboxylated OCN crossed the BBB and improved memory and learning while preventing anxiety and depression, whereas the carboxylated form of OCN passed through the BBB less efficiently.[167] OCN has also recently been shown to negatively impact the differentiation of oligodendrocytes and the production of myelin protein and blocking OCN activity through antibody neutralization resulted in up-regulation of myelin proteins.[168] OCN represents one of the powerful bone-derived osteokines that may freely cross the BBB in some forms and affect neuronal function.
2. Lipocalin2 (LCN2)
LCN2 is a glycoprotein that systemically regulates several functions, including bacterial defense, iron regulation, inflammation, regulation of cell death/cell survival, and even appetite regulation.[169,170] Approximately 50% of circulating LCN2 is produced and released by osteoblasts, and this fraction crosses the BBB to act in the hypothalamus.[171] By binding to melanocortin 4 receptors in the PVN, LCN2 exerts its neurologic effects to modulate systemic metabolism through melanocortin signaling impacting obesity and diabetes.[172] In the context of neurodegenerative diseases like AD, LCN2 may aggravate disease by increasing neuron vulnerability to Aβ and inhibit the neuroprotective effects of tumor necrosis factor (TNF) receptor 2.[173] In addition, LCN2 demonstrates a negative synergistic effect that aggravates the neurotoxicity of Aβ, TNF-α, or lipopolysaccharides.[174] LCN2 both aggravates and alleviates the causes and symptoms of its metabolic outcomes and aggravates NDs, prompting further study to fully elucidate its mechanisms of action. Nonetheless, LCN2 represents another bone-derived factor that crosses the BBB and influences neurologic function.
3. SOST
SOST is an osteokine produced solely by osteocytes and inhibits Wnt /β-catenin signaling by binding to lactate dehydrogenase receptor-related protein 4/5/6. It regulates bone formation by limiting osteoblast proliferation. Since the Wnt/β-catenin signaling pathway is essential for neurogenesis, synaptic plasticity, and BBB integrity in the brain and is associated with AD pathophysiology,[175] osteocyte-produced SOST may antagonize these functions in the brain should it or osteocyte-EVs containing this signaling factor cross the BBB as has been theorized in NDs due to increased BBB permeability.
4. DKK1
Like SOST, DKK1 is another Wnt signaling antagonist highly expressed by bone cells that regulate bone mass; however, DKK1 is expressed by some neurons within the brain. Excessive DKK1 signaling in the brain leads to synaptic decline and neuronal apoptosis, and DKK1 is overexpressed in the brains of AD patients and transgenic AD mice.[176] DKK1 in mice also impairs long-term potentiation, learning, and memory.[134] While a neural release of DKK1 may be responsible for the phenotypic findings involved in lost Wnt signaling in NDs, DKK1 may cross the BBB by direct transport via epithelial cells, by osteocytic EVs, and also potentially by crossing a damaged BBB in diseased brain tissues,[12] identifying DKK1 as a potential osteokine and member of the bone-brain signaling axis.
5. Fibroblast growth factor 23 (FGF23)
FGF23 is mostly expressed by bone osteoblasts and osteocytes, although it has been found in low levels in some brain areas, such as hypothalamus, hippocampus, and cortex and can be found in the cerebrospinal fluid.[177,178] Primarily, osteocyte-released, circulating FGF23 regulates phosphate homeostasis and transport in the kidney and vitamin D metabolism by modulating serum phosphate and vitamin D levels. Interestingly, the FGF family may cross the BBB, potentially extending to FGF23 as well.[179,180] While definitive roles for FGF23 in brain function have not been confirmed, FGF23 has emerged as a novel biomarker for vascular disease, including stroke and cerebral small vessel disease,[181,182] as well as a predictor for dementia in some patients, and impaired FGF23 signaling in mice leads to lost hippocampal function.[177] Although these conditions may be more directly related to hypertension through FGF23’s regulation of kidney function, the possibility of its role as a brain-targeting osteokine remains understudied.
6. OPN
OPN is a bone-derived proinflammatory glycoprotein that mediates osteo-immune crosstalk. In addition to bone cells, OPN is secreted by many cell types, including activated macrophages and T-lymphocytes.[183] In bones, OPN is present in the ECM and promotes bone resorption supporting bone demineralization by anchoring osteoclasts to the bone mineral matrix. In the brain, OPN is a key factor in tissue repair and ECM remodeling after ischemic stroke by regulating microglia activation. Interestingly, OPN is a regulator of neuron inflammation by inhibiting microglial superoxide production to increase cellular survival under stress conditions. OPN is also elevated in the serum and cerebrospinal fluid of AD patients and some brain regions of PD patients.[184,185] Interestingly, patients with high serum OPN levels often show low BMD.[186] The multifunctional osteokine OPN is found in bone and brain and in circulation, cementing its position within the bone-brain axis.
7. RANKL
RANKL, also known as TNF superfamily member 11, represents another secreted protein produced in bone and T-lymphocytes. RANKL binds its receptor expressed on osteoclasts and osteoclast precursors to activate osteoclast differentiation and increase bone resorption. In the brain, RANKL may signal to microglia, other resident macrophages, neurons, and oligodendrocyte precursor cells to combat neuroinflammation by decreasing the expression of inflammatory markers such as inducible nitric oxide synthase and cyclooxygenase, potentially by blocking expression of the Toll-like receptor adaptor proteins.[187,188] RANKL also helps to regulate body temperature and fever responses through action in the hypothalamus.[189] Interestingly, treatment of mice with an anti-RANKL antibody seemed to ameliorate symptoms of chronic stress and depression-like symptoms in mice.[190] Further studies are needed to elucidate the precise role of bone-derived RANKL signaling in the brain, but evidence supports its role as a potential osteokine.
8. BMPs
BMPs are a wide class of growth factors that belong to the largest subfamily of the TGF-β superfamily and are expressed in many tissues throughout the body. BMPs are sequestered in the ECM of their host tissues during deposition and development.[191,192] In bone, BMPs play a dynamic osteoinductive role in bone metabolism as skeletal tissue is consistently turned-over during bone remodeling cycling the deposition and release of active BMPs within the bone ECM. In the brain, BMPs play a large role during development in cell fate decisions and patterning of neural stem cells, adult neuron and glial cell maintenance, and neural stem cell proliferation.[193,194] BMP signaling in the hypothalamus also plays roles in thermoregulation and appetite control. Given their osteoinductive and neurogenerative effects, BMPs may be avenues for treatment for both skeletal and neurodegenerative diseases.
9. IGF-1
IGF-1 is a hormone structurally similar to insulin, primarily synthesized in liver, but also produced by bone and brain cells. IGF-1 signaling is central to pathways that promote cell growth and survival, maturation, and proliferation, allowing for tissue growth and renewal. In bone, IGF-1 regulates bone length of long limb bones by affecting cells within the growth plate. Overexpression of IGF-1 in osteoblasts increased bone turnover and cortical bone mass and size, and losses in IGF-1 with age are associated with low BMD and osteoporosis.[195] In the brain, IGF-1 is involved in neurodevelopment prenatally and in the early post-natal period, as well as in plasticity and remodeling throughout adult life. IGF-1 may be involved in the progression of AD by increasing the susceptibility of inflammation in response to Aβ plaque toxicity. Interestingly, the pathologies related to Aβ neuroinflammation and loss of spatial memory were alleviated in neuron-specific IGF1 receptor-null mice.[196,197] Further research is required to ascertain if IGF-1 is involved in bone-brain tissue crosstalk as part of the bone-brain axis.
PROTEOMIC WORKFLOWS AND STUDIES FOR BONE BIOLOGY
High-resolution mass spectrometry-based proteomics techniques have gained increasing popularity in biological studies due to their high throughput, sensitivity, and reproducibility for the study of proteins involved in biological and macromolecular processes. Technological and computational advancements over the past decades have greatly improved mass spectrometry-based proteomic workflows to profile thousands of proteins in human tissues and model systems.[1,198] Mass spectrometric analysis is useful to determine quantitative changes when comparing different conditions or disease states in biologic samples. For example, it has been found that cellular senescence and inflammation are key drivers of aging in the skeleton and the brain independently and that combating these specific cellular mechanisms in these tissues can defend against osteoporosis, OA, and even neurodegeneration.[199–202] Mass spectrometric approaches have been key in identifying protein biomarkers of cellular senescent phenotypes,[203] and utilizing these techniques within the bone-brain axis provides a unique opportunity to identify novel biomarkers for both brain and bone diseases, to discover new important regulatory members of this signaling axis, and to generate possible novel treatments for these degenerative diseases. Proteins identified in bone and brain tissue or the circulation may act as long-distance signaling molecules and demonstrate bone-brain communication in both directions. Given the power and potential of proteomic approaches to aid in the study of macromolecular signaling and tissue crosstalk, we will focus here on recent advances in quantitative mass spectrometric workflows and studies for brain and bone tissues to motivate its use in the study of the bone-brain axis.
The use of mass spectrometry to study molecular signaling in brain is very popular, while the study of molecular signaling in bone has had limited success primarily due to the difficulties involved in sample preparation to isolate proteins from the mineralized bone matrix that is dominated by structural proteins. In recent studies focused on specific brain tissues, scientists have been able to routinely quantify 2,000 to 5,000 or even more individual proteins, even deciphering the sub-proteomes from different brain areas.[198,204,205] Recent protein extraction approaches for mass spectrometry in bone and other skeletal tissues are summarized in Table 2. Studies could be divided into protocols that contained a bone demineralization step prior to protein extraction, and others that used the bone as was (without bone mineralization) for protein extraction. Protein extraction protocols that did not remove the mineralized bone matrix typically resulted in the identification of tens to hundreds of identified and quantified proteins.[206–210] However, for several studies, the optimizations in sample preparation, such as demineralization of the bone matrix from samples using hydrochloric acid followed by subsequent protein extraction with guanidine-hydrochloride and/or ethylenediaminetetraacetic acid (EDTA) before protein digestion, resulted in identification and quantification of thousands of proteins.[2,211–214]
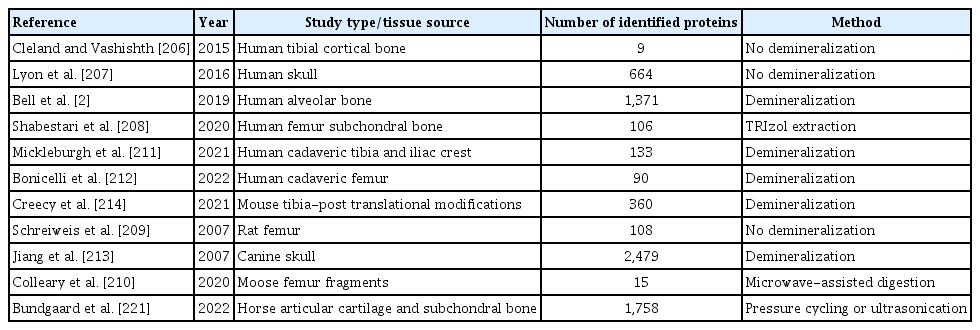
Recent mass spectrometry approaches for bone tissue
In 2007, Jiang et al.[213] demonstrated the demineralization of the bone matrix in canine skull with 1.2 M hydrochloric acid followed by protein extraction with guanidine-hydrochloride and EDTA for 72 hr yielded mass spectrometric identification of 2,479 proteins. Specifically, Jiang et al.[213] identified numerous bone-specific proteins associated with bone formation and resorption from canine skulls including OCN, osteonectin, and bone sialoprotein 2 (IBSP). ECM proteins have been implicated in disease progression, and changes in ECM proteins are significant in determining cell fate through mechanical interactions with the stroma. Collagens, collagenases, matrix metalloproteinases, and matrix glia-protein were among the multiple ECM proteins identified by Jiang et al.[213]. The identification of bone-specific and ECM proteins after demineralization, protein extraction, and proteolytic digestion represent a pathway toward understanding cellular mechanisms underlying bone biology and bone disease.
Data-independent acquisition (DIA) was established as a label-free mass spectrometry approach to increase the number of identified and quantified proteins in a highly quantitative and accurate fashion, avoiding so-called ‘missing values’.[215–218] DIA approaches to measure and quantify all detectable peptides to provide the most comprehensive mass spectral dataset for a sample cohort. DIA is performed by sequentially isolating peptide precursor ions from ‘mass to charge’ (m/z) windows, rather than isolating each precursor ion individually. For each ‘cycle’ of typically 2 to 3 sec, precursor ions from each of the m/z windows are sequentially fragmented to obtain complex MS/MS spectra, and the MS1 windows within each cycle will span the entire MS1 mass range. The complex MS/MS spectra are subsequently analyzed with various computational strategies using publicly available reference spectral libraries, such as pan-human libraries,[219] custom-generated spectral libraries, or applying spectral library-free workflows.[220]
A high-throughput protocol developed by Bundgaard et al.[221] used a method applying 60 min of pressure cycling or 30 min of ultrasonication in guanidine-hydrochloride to remove the mineralized bone matrix before proteolytic digestion. This resulted in the identification and quantitation of 1,758 proteins. This study mapped the articular cartilage and subchondral bone proteomes of horses using DIA quantification to investigate the changes in molecular events within the osteochondral unit in OA. Proteins identified only in subchondral bone regions included the bone-specific proteins osteomodulin, OCN, secreted protein acidic and rich in cysteine (SPARC), and BMPs, highlighting the specificity of the extraction and analysis protocols for bone samples. ECM proteins identified from horse articular cartilage and subchondral bone samples include broader proteins including collagens, matrix metalloproteinases, decorin and lumican. The signaling proteins TGF-β1 and IGF1, known to be involved in homeostatic and disease processes in each individual tissue, were identified in both bone and cartilage regions. Bundgaard et al.[221] demonstrated the utility of DIA-mass spectrometry for identification and quantitation of bone and cartilage proteins and provided a horse-specific spectral library of these proteins for future studies.
The comprehensive label-free quantification workflow DIA-mass spectrometry was also used in a recent study by Rose et al.[222], who demonstrated the identification and quantitation of 2,108 proteins from mouse femurs by adapting the demineralization/extraction method from Jiang et al.[213] and combining it with the very comprehensive and accurate DIA-mass spectrometry quantification. Proteins identified from mouse femur bones in this study included those that play key roles in bone health, such as several ECM proteins, bone-specific signaling molecules, and proteins associated with the senescence-associated secretory phenotype.[203] ECM proteins identified by Rose et al.[222] featured many collagen subtypes, and the quantitative distribution was equivalent to the abundance of each collagen family in bone with a predominance of fibril-forming collagens, such as Col1a1 and Col1a2 subchains. Collagen-modifying enzymes important in collagen formation and maturation included prolyl 3 hydroxylase 3 and lysyl oxidase homolog 2, and these were also found to be highly abundant within the bone ECM. Bone-specific proteins that play a role in bone formation and resorption, which are functionally associated with osteoblasts, osteoclasts, and osteocytes were identified as well including IBSP, SPARC, OPN, matrix extracellular phosphoglycoprotein, and tartrate-resistant acid phosphatase type 5. TGF-β2, a signaling protein that is involved in brain and bone functions as previously described, was also identified. The report by Rose et al.[222] provides a comprehensive landscape of the proteins involved in bone health and disease progression while demonstrating the utility of DIA-mass spectrometry for simultaneous identification and quantification of these proteins.
A comparison of high-quality proteome data sets from the bone and the brain of model organisms shows surprising amounts of similarity implicating disease processes. A comparison of the proteome datasets from Rose et al.[222], who investigated mouse femoral samples, and from Koopmans et al.[205], who analyzed the hippocampal synaptosomes in mice and other organisms, demonstrates the importance of utilizing proteomics to study the bone-brain axis. Both studies generated a comparable large number of proteins with >2,000 each from mouse bone and mouse brain, respectively. Within these datasets, 910 identified proteins were found in both the mouse bone and mouse hippocampal proteomes accounting for roughly 40% of proteins in each independent study (Fig. 5A). The Kyoto Encyclopedia of Genes and Genomes pathway analysis of these overlapping proteins showed significant enrichments in metabolic pathways and for many disease processes such as AD, PD, and HD (Fig. 5B). Visualization of the overlap in bone and brain by the pathway Enrichment Score shows a much more complex relationship between the 2 organ systems. Interestingly, the pathway representation is clearly dominated by neurodegenerative processes and many other biologic functions, such as oxidative phosphorylation, the citric acid cycle, protein processing, and the proteasome complex (Fig. 5C). The identification of these disease processes and shared functions in bone and brain proteomes in mice signify the need to understand the biologic mechanisms behind these disease and functional processes in each tissue and how they may impact the health of the reciprocal tissue. In this presented comparison, the startling overabundance of neurodegenerative disease processes found in common in both tissues highlights the further need to study the bone-brain axis, while proteomic technologies demonstrate their use as powerful tools in discovery and hypothesis-driven science.
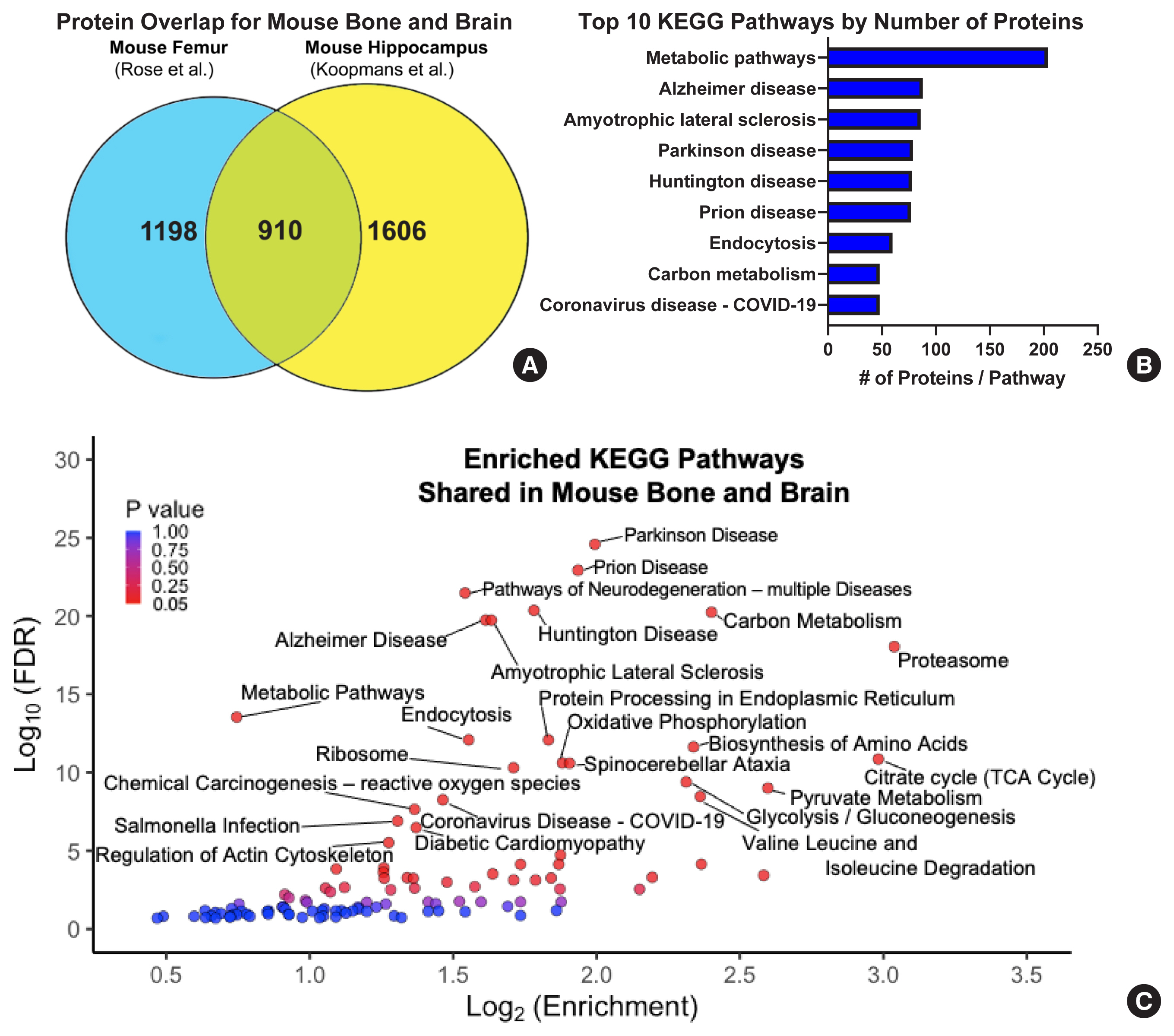
Example comparison of proteomes from the bone and brain of mice identifies important degenerative disease processes. (A) Comparison of high-quality proteome datasets identified from the mouse femur and from the hippocampal synaptosome show a large amount of similarity with an overlap accounting for nearly 40% of each individual dataset. (B) Ranking of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways by the number of proteins from the overlap in each study showed between 50–200+ individual proteins in each enriched pathway for the top 10 pathways by protein count. (C) Volcano plot of ‘enrichment scores’ for the identified KEGG pathways of the 910 shared proteins in mouse bone and brain demonstrating an overrepresentation for neurodegenerative disease processes and other important cellular metabolic functions. An false discovery rate (FDR) cut-off of −log10 (FDR)>5 was used to determine pathway annotation. Enrichment, the number of observed genes divided by the number of expected genes from each KEGG category in the 910-gene list (according to Database for Annotation, Visualization and Integrated Discovery).
The studies highlighted above demonstrated preparation methods of bone samples that allow for excellent depth of coverage in proteomic experiments of animal models. The protocols from Jiang et al.[213], Bundgaard et al.[221] and Rose et al.[222] retain information about bone-specific, ECM, and signaling proteins, allowing comparisons to be made between bone and brain proteomes in multiple model systems. As proteomic studies of mineralized or other highly complex tissues gain more popularity and protocols provide consistent and reliable extraction of biologically relevant proteins, the field is now set up to shift to more studies involving human bone tissues. To date, the most efficient and comprehensive protocols for bone tissues have been applied to animal model tissues, including those discussed above (rat, horse, mouse), and while these are important in the demonstration of the feasibility of extraction protocols and for the discovery of novel biology utilizing animal models, the translation of these studies to human subjects remain rare. The focus on animal models limits clinical translation due to the differences in basic biology and the difficulty of finding homology among peptide sequences across multiple model systems and human tissues. Tools and pipelines are starting to be implemented for future human bone studies, specifically tissue preparation with skilled clinical partners that are directly involved in attaining and preparing human biopsies, including removal of cartilage and preservation of bone tissues for successful proteomic studies.
The ability for mass spectrometric workflows to separate, identify, and quantify low-abundant signaling molecules from the bone, brain, and circulation can help elucidate biomarkers for bone and brain diseases. The presence of specific signaling factors, as well as those not yet identified, in the skeleton from the nervous system may be ascertained through careful sample preparation that considers the specific challenges presented by mineralized bone tissue and DIA methods. Identification of molecular markers that signal between the bone and the brain in circulation by mass spectrometry will also help more fully describe the mechanisms of signaling between these 2 organ systems in homeostasis and disease. Given the power and potential of proteomics approaches to aid in the study of macromolecular signaling and tissue crosstalk, mass spectrometry presents itself as an ideal tool for the study of the bone-brain axis, especially in clinically relevant human samples.
CONCLUSIONS
The bone-brain axis has been observed for decades. Reaching back to the original observations of paradoxical accelerated bone healing in TBI patients from as early as the 1960’s to 1970’s, clinical evidence has mounted that demands further mechanistic studies into the relationship between the skeleton and the nervous system. The current body of literature demonstrates the similarities in form and function for several different cell types in bone and brain that have loose orthologs to one another. Intriguingly, these cell types respond to and utilize several of the same molecular signaling mechanisms that may be leveraged in future studies and treatments. In future clinical approaches, the broad classes of neurokines and osteokines that modulate the activities in one organ may directly impact the health and function on the other end of the bone-brain axis. While transcriptomic-based studies have shown that several markers of bone cells may be expressed and act in the brain, and vice versa, to trace molecular signaling, proteomic approaches are necessary to elucidate specific molecular crosstalk in circulation between these seemingly distal organs.
Acknowledgments
We acknowledge support from the National Institutes of Health (NIH): specifically the National Institute on Aging: U01 AG060906 (PI: Schilling), P01 AG066591 (PI: Campisi/Ellerby) and T32 AG000266 to Schurman (PI: Campisi/Ellerby). We also thank the National Institute of Dental and Craniofacial Research (NIDCR): R01 DE019284 (PI: Alliston), and the Office of the Director (OD) S10 OD028654 (PI: Schilling). Dr. Schilling is grateful for support from the Forever Healthy Foundation.
Notes
Ethics approval and consent to participate
Not applicable.
Conflict of interest
No potential conflict of interest relevant to this article was reported.