Anti-Müllerian Hormone Negatively Regulates Osteoclast Differentiation by Suppressing the Receptor Activator of Nuclear Factor-κB Ligand Pathway
Article information

Abstract
Background
Multiple members of the transforming growth factor-β (TGF-β) superfamily have well-established roles in bone homeostasis. Anti-Müllerian hormone (AMH) is a member of TGF-β superfamily of glycoproteins that is responsible for the regression of fetal Müllerian ducts and the transcription inhibition of gonadal steroidogenic enzymes. However, the involvement of AMH in bone remodeling is unknown. Therefore, we investigated whether AMH has an effect on bone cells as other TGF-β superfamily members do.
Methods
To identify the roles of AMH in bone cells, we administered AMH during osteoblast and osteoclast differentiation, cultured the cells, and then stained the cultured cells with Alizarin red and tartrate-resistant acid phosphatase, respectively. We analyzed the expression of osteoblast- or osteoclast-related genes using real-time polymerase chain reaction and western blot.
Results
AMH does not affect bone morphogenetic protein 2-mediated osteoblast differentiation but inhibits receptor activator of nuclear factor-κB (NF-κB) ligand-induced osteoclast differentiation. The inhibitory effect of AMH on osteoclast differentiation is mediated by IκB-NF-κB signaling.
Conclusions
AMH negatively regulates osteoclast differentiation without affecting osteoblast differentiation.
INTRODUCTION
The balance between bone-resorbing osteoclasts and bone-forming osteoblasts is essential for the maintenance of bone homeostasis.[1] The imbalance in these bone cells’ activity causes various bone diseases such as osteoporosis, rheumatoid arthritis, paget’s disease, and osteopetrosis.[2] Osteoporosis is characterized by low bone mass, micro-architectural deterioration of bone tissue and consequent increase in fracture risk is caused by excessive bone resorption relative to bone formation.[3,4] While osteopetrosis is characterized by sclerotic and thick but weak and brittle bones is caused by the failure of osteoclasts to resorb bone.[5]
Multiple hormones, growth factors, and cytokines are involved in bone remodeling through regulating the proliferation, differentiation, and activation of bone cells as well as the communication between them.[6,7] In particular, the growth factors belonging to the transforming growth factor β (TGF-β) superfamily play an important role in the balance between bone formation and resorption through directly or indirectly regulating osteogenesis and osteoclastogenesis.[6,8] There are several subfamilies of TGF-β superfamily including the TGF-β, Nodal, Activin subfamily, the bone morphogenetic protein (BMP), growth/differentiation factor (GDF) family, and the group of anti-Müllerian hormone/Müllerian inhibiting substance (AMH/MIS).[6,8]
TGF-β/BMPs have absolutely recognized roles in bone formation both in vivo and in vitro.[6,8,9] Especially, BMP-2, BMP-4, BMP-6, and BMP-9 induce osteoblastic differentiation and bone formation, in addition, BMP-2 and BMP-7 in combination further enhance osteoblastic differentiation than that induced by BMP-2 or BMP-7 alone.[10–15] TGF-β1 also drastically enhances ectopic bone formation induced by BMP-2.[15] The roles of TGF-β/BMPs in bone resorption are relatively less established compared to bone formation and still controversial. BMP-7, and BMP-9 promote receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL)-induced osteoclast differentiation.[16,17] While BMP-2, TGF-β1 and TGF-β2 have a biphasic effect on osteoclast differentiation concentration- or stage of differentiation-dependently.[18–20]
AMH, also known as MIS, is a dimeric glycoprotein exclusively expressed by granulosa cells of pre-antral and small antral follicles in the ovary. AMH plays an important role in chronic anovulation by inhibiting the initial and cyclic recruitment of ovarian follicles and by promoting follicular arrest.[21,22] In addition, AMH has antiproliferative effects on ovarian and breast tumor cell lines.[21,23] Since AMH is a distinct member of the TGF-β superfamily, it is expected that AMH signaling is similar to that of the well-defined signaling pathway of other TGF-β family members.[21] However, despite most family members of TGF-β superfamily are involved in a vast majority of cellular processes and thereby exhibiting versatile functions in the body, the known function of AMH is still restricted to the reproductive organs.[6,21,24] In the present study, we investigated whether 2 types of bone cells are target cell(s) to translate the signals of AMH, and AMH regulates osteogenesis and osteoclastogenesis like other TGF-β family members.
METHODS
1. Reagents
Recombinant human RANKL was purified from bacteria. Recombinant human macrophage colony-stimulating factor (M-CSF) was a gift from Dr. Daved Fremont (Washington University, St. Louis, MO, USA). Recombinant human BMP2 protein was purchased from Cowellmedi (Busan, Korea). Ascorbic acid was purchased from Junsei Chemical (Tokyo, Japan). β-glycerophosphate was purchased from Sigma-Aldrich (St. Louis, MO, USA). Recombinant mouse AMH was purchased from Cloud-Clone Crop. (Wuhan, China).
2. Osteoblast differentiation
Primary osteoblast precursor cells were isolated from newborn of ICR mouse calvaria via enzymatic digestion with 0.1% collagenase (Life Technologies, Carlsbad, CA, USA) and 0.2% dispase II (Roche Diagnostics, Mannheim, Germany). Primary osteoblast precursor cells were cultured in an osteogenic medium containing BMP2 (100 ng/mL; Cowellmedi), ascorbic acid (50 μg/mL; Junsei Chemical), and β-glycerophosphate (100 mM; Sigma-Aldrich) for 3 or 6 days. Cultured cells for 3 days were lysed using osteoblast lysis buffer (50 mM Tris-HCl [pH 7.4], 1% Triton X-100, 150 mM NaCl, and 1 mM ethylenediaminetetraacetic acid [EDTA]). Cell lysates were incubated with p-nitrophenyl phosphate substrate (Sigma-Aldrich), and alkaline phosphatase (ALP) activity was determined by measuring the absorbance at 405 nm using a spectrophotometer. Cultured cells for 6 days were fixed with 70% ethanol and stained with 40 mM Alizarin red (pH 4.2). After removing nonspecific staining with phosphate buffered saline, stained cells were visualized with a CanoScan 4400F (Canon Inc., Tokyo, Japan). Alizarin red-stained cells were incubated with 10% cetylpyridinium chloride solution for 30 min at room temperature, and the absorbance was determined at 562 nm.
3. Osteoclast differentiation
Mouse bone marrow cells were isolated from tibiae and femurs of 6-week-old ICR mice by flushing the bone marrow with α-minimal essential medium (α-MEM; Hyclone Lab Inc., Logan, UT, USA). Bone marrow cells were cultured for 3 days in α-MEM (Hyclone Lab Inc.) containing 10% fetal bovine serum (FBS; Hyclone Lab Inc.) in the presence of M-CSF (30 ng/mL). Adherent cells were further cultured in the presence of M-CSF (30 ng/mL) and RANKL (150 ng/mL) for 3 days. Cultured cells were fixed with 3.7% formalin and stained for tartrate-resistant acid phosphatase (TRAP). TRAP-positive cells with more than 3 nuclei were considered TRAP-positive MNCs. Cells were observed using a Leica DMIRB microscope equipped with an N Plan 10×0.25 numerical aperture objective lens (Leica Microsystems, Wetzlar, Germany). Images were captured with a ProgRes CFscan camera (Jenoptik, Jena, Germany) using ProgRes Capture Pro software (Jenoptik).
4. The proliferation of osteoclast precursor cells
Osteoclast precursor cells were cultured for 3 days in α-MEM (Hyclone Lab Inc.) containing 10% FBS (Hyclone Lab Inc.) in the presence of M-CSF (30 ng/mL) in a 96-well plate. The cell proliferation was measured with the MTT kit (Sigma-Aldrich) according to the manufacturer’s protocol.
5. Real-time polymerase chain reaction (PCR)
Total RNA was prepared using the Qiazol reagent (Qiagen, Valencia, CA, USA), and 2 μg of the isolated RNA was reverse transcribed into cDNA using Superscript II Reverse Transcriptase (Invitrogen; Thermo Fisher Scientific, lnc., Waltham, MA, USA). Quantitative real-time PCR analysis was performed in triplicate with a Rotor-Gene Q (Qiagen) with SYBR Green (Qiagen). Expression levels were normalized against endogenous glyceraldehyde 3-phosphate dehydrogenase levels. The following primers were used: Gadph, 5′-TGA CCA CAG TCC ATG CCA TCA CTG-3′ and 5′-CAG GAG ACA ACC TGG TCC TCA GTG-3′; cfos, 5′-ATG GGC TCT CCT GTC AAC ACA-3′ and 5′-TGG CAA TCT CAG TCT GCA ACG CAG-3′; Nfatc1, 5′-CTC GAA AGA CAG CAC TGG AGC AT-3′ and 5′-CGG CTG CCT TCC GTC TCA TAG-3′; Acp5, 5′-CTG GAG TGC ACG ATG CCA GCG ACA-3′ and 5′-TCC GTG CTC GGC GAT GGA CCA GA-3′; Runx2, 5′-CCC AGC CAC CTT TAC CTA CA-3′ and 5′-CAG CGT CAA CAC CAT TC-3′; Alpl, 5′-CAA GGA TAT CGA CGT GAT CAT G-3′ and 5′-GTC AGT CAG GTT CCG ATT C-3′; Ibsp, 5′-GGA AGA GGA GAC TTC AAA CGA AG-3′ and 5′-CAT CCA CTT CTG CTT CGT TC-3′; Bglap, 5′-ATG AGG ACC CTC TCT CTG CTC AC-3′ and 5′-AGA GCA AAC TGC AGA AGC TGA GAG-3′; and Amh, 5′-GGA GTC TGC AGC ACT GAC TC-3 and 5′-TCA CTT CAG CCA GAT GTA GG-3′.
6. Western blot
Cultured cells were lysed in extraction buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 1 mM PMSF, and protease inhibitor cocktail). After centrifugation, equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). Membranes were incubated with the appropriate antibodies against c-Fos, NFATc1, Cathepsin K, Actin (Santa Cruz Biotechnology, Dallas, TX, USA), phospho-p38, p38, phospho-c-JUN N-terminal kinase (JNK), JNK, phospho-Erk, Erk, and IκB (Cell Signaling Technology, Danvers, MA, USA). Signals were detected with an enhanced chemiluminescence reagent (Millipore) and analyzed using an Azure c300 luminescent image analyzer (Azure Biosystems, Dublin, CA, USA).
7. Statistical analyses
All values are expressed as the means±standard deviation. Statistical significance was determined by using 2-tailed Student’s t-tests for 2 independent samples or analysis of variance with post-hoc Tukey’s honestly significant difference test for multiple group comparisons. A P-value of less than 0.05 was considered statistically significant.
RESULTS
1. AMH is not expressed in both osteoblasts and osteoclasts
To determine whether AMH participates in osteoblast differentiation, gene expression of Amh was analyzed during the differentiation of osteoblasts. Primary osteoblast precursor cells were induced to osteoblasts by culturing in an osteogenic medium containing BMP-2, ascorbic acid, and β-glycerophosphate. It was observed that Amh gene expression was not detected in BMP-2-induced osteoblast differentiation while there were inductions of osteoblast differentiation marker genes, including Runx2, Alpl, Ibsp, and Bglap (Fig. 1A). Next, Amh gene expression was analyzed during osteoclast differentiation. Bone marrow-derived macrophages (BMMs) were differentiated to osteoclast by culturing in an osteoclastogenic medium containing M-CSF and RANKL. Despite induction of osteoclast differentiation marker genes such as c-fos, Nfatc1, and Acp5, Amh gene expression was undetectable in RANKL-induced osteoclast differentiation (Fig. 1B).

Expression of Anti-Müllerian hormone (AMH) in bone cells. (A) Osteoblasts were cultured with osteogenic medium containing bone morphogenetic proteins 2, ascorbic acid, and β-glycerophosphate for the indicated times. (B) Bone marrow-derived macrophages were cultured with macrophage colony-stimulating factor (M-CSF) alone or M-CSF and receptor activator of nuclear factor-κB ligand (RANKL) for the indicated times. (A, B) Total RNA was extracted at each indicated time point. Quantitative real-time polymerase chain reaction analysis was performed to measure the mRNA expression levels of the target genes. #P<0.05. *P<0.01. **P<0.001 vs. control.
2. AMH does not affect BMP2-induced osteoblast differentiation
Since Amh was not expressed during osteoblast differentiation, the effect of AMH on osteoblast differentiation and function via treatment of exogeneous AMH during the BMP2-induced osteoblast differentiation of primary osteoblast precursor cells. Upon treatment of exogeneous AMH, there was no significant difference in ALP activity and nodule formation was slightly increased (Fig. 2A, B). Furthermore, AMH did not have any effect on gene expressions of osteoblast differentiation marker genes such as Runx2, Alpl, Ibsp, and Bglap (Fig. 2C). These results indicate that AMH is not involved in BMP2-induced osteoblast differentiation of primary osteoblast precursor cells.
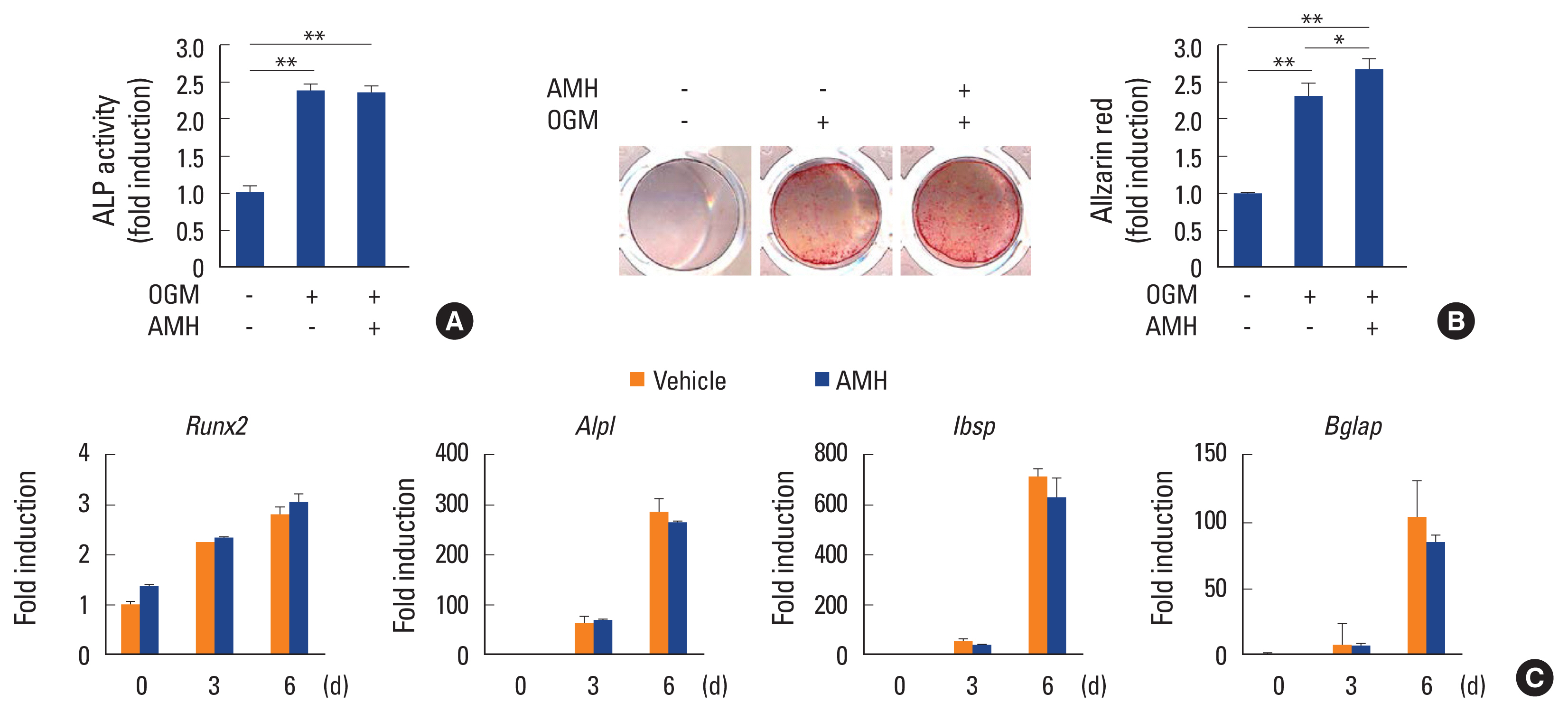
Anti-Müllerian hormone (AMH) does not affect osteoblast differentiation. (A–C) Osteoblasts were cultured with osteogenic medium (OGM) in the absence or presence of AMH (100 ng/mL). (A) Cells were cultured for 3 days and subjected to alkaline phosphatase (ALP) activity assay. (B) Cultured cells for 6 days were fixed and stained with Alizarin red (left panel). Staining intensities were quantified via densitometry at 562 nm (right panel). (C) Total RNA was extracted at each indicated time point. Quantitative real-time polymerase chain reaction analysis was performed to measure the mRNA expression levels of the target genes. *P<0.01. **P<0.001 vs. control.
3. AMH inhibits RANKL-induced osteoclast differentiation
Next, the effect of AMH on RANKL-induced osteoclast differentiation of BMMs was investigated via treatment of AMH in various doses during the osteoclast differentiation. AMH treatment significantly inhibited TRAP-positive multinuclear osteoclast formation in a dose dependent manner according to TRAP stain analyses without affecting proliferation of osteoclast precursor cells (Fig. 3A). Furthermore, AMH dramatically suppressed expression levels of osteoclast differentiation markers such as c-Fos, NFATc1, and TRAP at both mRNA and protein levels (Fig. 3B, C). Since AMH inhibited osteoclast differentiation, its effect on early RANKL signaling pathway was investigated. Phosphorylation of p38 and Erk was increased by AMH pretreatment, which seems to be activated by AMH rather than affecting the signaling activated by RANKL. Among several signaling pathways activated by RANKL, JNK activation was inhibited, and above all, IκB degradation was most strongly inhibited (Fig. 4). These results suggest that the strong inhibition of IκB degradation which occur simultaneously with inhibition of JNK activation is responsible for inhibitory effect of AMH on RANKL-induced osteoclast differentiation.
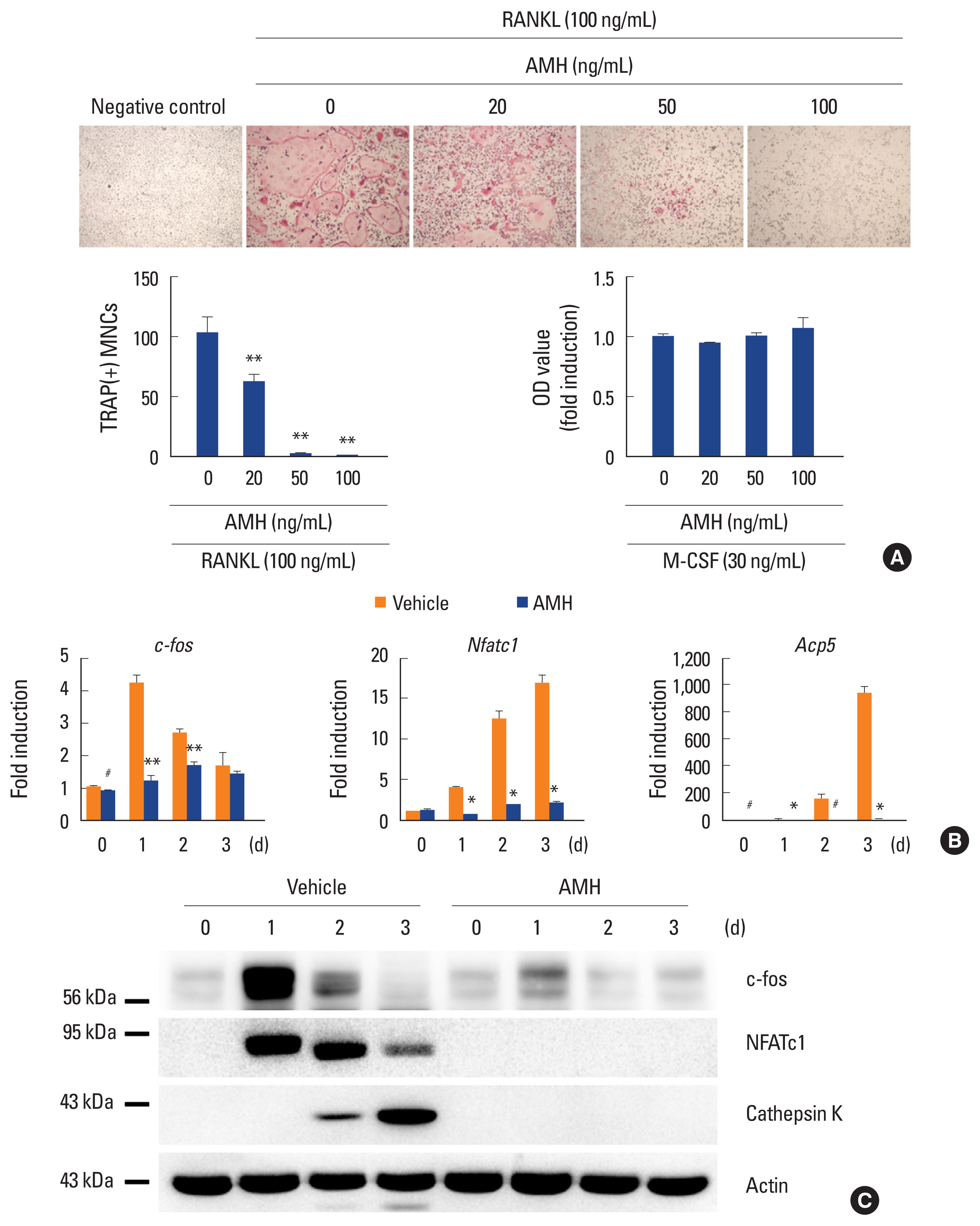
Anti-Müllerian hormone (AMH) inhibits osteoclast differentiation. (A–C) Bone marrow-derived macrophages were cultured with macrophage colony-stimulating factor (M-CSF) alone or M-CSF and receptor activator of nuclear factor-κB ligand (RANKL) in the absence or presence of AMH as indicated. (A) Cultured cells were fixed and stained for TRAP (upper panel). Numbers of TRAP (+) multinucleated cells were counted (lower left panel). The cell proliferation was measured by MTT assay (lower right panel). (B) Total RNA was extracted at each indicated time point. Quantitative real-time polymerase chain reaction analysis was performed to measure the mRNA expression levels of the target genes. (C) Whole cell lysates were analyzed via western blotting using specific antibodies as indicated. #P<0.05. *P<0.01. **P<0.001 vs. control.
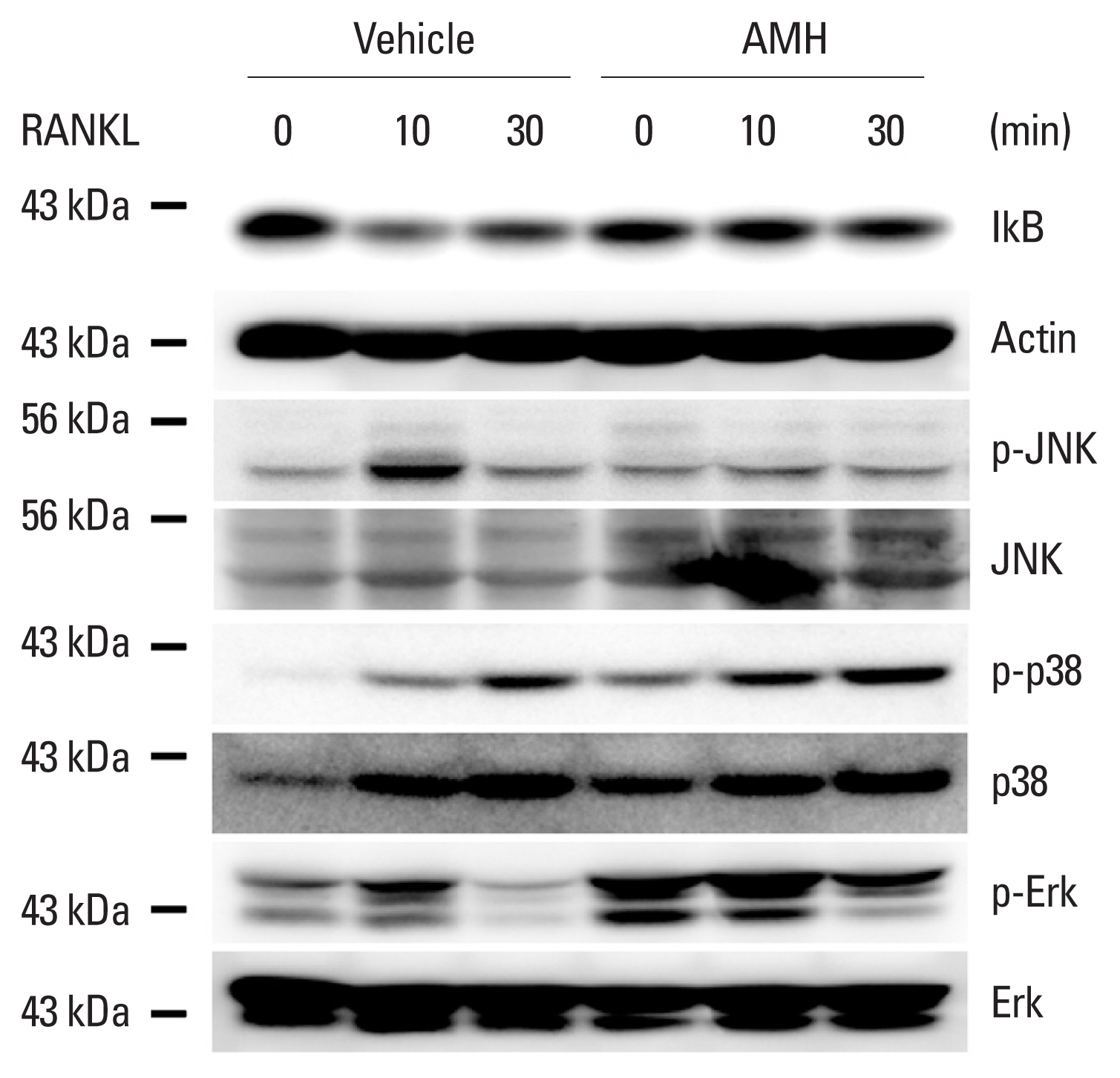
Anti-Müllerian hormone (AMH) inhibits receptor activator of nuclear factor-κB ligand (RANKL)-induced IkB degradation. Bone marrow-derived macrophages were serum-starved, pretreated with vehicle or AMH for 1 hr and stimulated with RANKL (500 ng/mL) for the indicated times. Whole cell lysates were analyzed via western blotting using specific antibodies as indicated.
DISCUSSION
AMH has been demonstrated that plays an important role in ovarian function.[22] In addition to AMH, BMPs is known for its critical role in the regulation of ovarian function, of which BMP-2, BMP-6, BMP-7, BMP-15, and GDF-9 are expressed in the human ovary, and specially, BMP-15 and GDF-9 are exclusively expressed in the oocytes.[25–29] Ogura-Nose et al. [25] showed that BMP-2, BMP-6, BMP-7, and BMP-15, but not GDF-9, induced both mRNA and protein expression of AMH in human granulosa cells. As other members of the TGF-β family, AMH first transmits signals through the formation of heteromeric complexes of specific type I and type II serine/threonine kinase receptors.[21] The activation of specific type II receptor by binding AMH is followed by phosphorylation of the type I receptor. Activated type I receptor results in phosphorylation of the downstream Smad proteins. Phosphorylated downstream Smad proteins form a complex with common Smad4 and this complex is translocated to the nucleus to regulate target gene expression.[21,30] AMH has been reported to activate BMP like a pathway through BMP type I receptors such as activin-like kinase (ALK)2 and ALK6 to activate the Smad5 and Smad1 pathways respectively.[30–32] The results imply that AMH expression is increased by BMPs and the Smad1 and Smad2 pathway are shared by the BMPs and AMH led us to test whether AMH is induced by BMP2 in osteoblasts and AMH can regulate osteoblast differentiation by activating BMP like pathway. Unexpectedly, AMH expression was not increased by BMP-2, nor AMH affect BMP-2-mediated osteoblast differentiation in primary osteoblasts indicating that osteoblasts are not target cells of AMH under the physiological condition.
AMH seems to activate the BMP-like pathway to induce Smad1-dependent signaling in osteoclast precursor cells. Josso et al. [33] reported that AMH type II receptor (AMHR-II) interacts with ALK5 and ALK6 among the type I receptors and interaction with ALK5 is ligand-independent while interaction with ALK6 is increased by AMH. They also showed that AMH activates Smad1 but not Smad2 signal transduction. Smad1 signal has been demonstrated to inhibit osteoclast differentiation. Inhibitory effect of TGF-β1 during human osteoclastogenesis is mediated by Smad1 signaling.[33] In addition, BMP-Smad1 signaling inhibits the initiation of osteoclast differentiation by suppressing the RANKL-NF-κB pathway.[20] Our results showed that AMH inhibited RANKL-induced osteoclast differentiation without affecting proliferation of osteoclast precursor cells and AMH pretreatment strongly caused a decrease in IκBα degradation medicated by RANKL. These results are consistent with that BMP2 pretreatment caused an increase in IκB and a decrease in NF-κB during M-CSF/RANKL-induced osteoclast commitment and indicating that AMH negatively regulates osteoclast differentiation by suppressing the RANKL-NF-κB pathway.
In this study, we have provided clues on the role of AMH during bone remodeling. AMH was found to inhibit osteoclast differentiation without affecting osteoblast differentiation. Further study on the role and action mechanism of AMH in osteoclasts and osteoblasts will help to understand the complex mechanisms that how TGF-β superfamily influence bone homeostasis and bone disease.
Notes
Funding
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (2019R1A5A2027521).
Ethics approval and consent to participate
This study conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved.
Conflict of interest
No potential conflict of interest relevant to this article was reported.