X-linked Hypophosphatemic Rickets, del(2)(q37.1;q37.3) Deletion Syndrome and Mosaic Turner Syndrome, mos 45,X/46,X, del(2)(q37.1;q37.3) in a 3-year-old Female
Article information
Abstract
There are currently no published cases that report concomitant Turner syndrome (TS), 2q37 deletion syndrome and X-linked hypophosphatemic rickets (XLH). Interestingly, since the clinical phenotypes of TS and 2q37 deletion syndrome overlap, the correct diagnosis may be missed without a standardized approach to genetic testing consisting of both karyotype and microarray. Both chromosome anomalies have been associated with short stature and a variety of skeletal abnormalities however to date no reports have associated these syndromes in association with a phosphate regulating endopeptidase homolog, X-linked (PHEX) gene deletion resulting in XLH. We report a 3-year-old female with 3 concurrent genetic disorders including a 9.98 Mb terminal deletion of chromosome 2: del(2)(q37.1;q37.3), XLH secondary to a small microdeletion of part of the PHEX gene, and mosaic TS (mos 45,X[32]/46,X[18]). This is the first case report of a patient with 2q37 deletion syndrome and mosaic TS (mos 45,X[32]/46,X[18]) found to have XLH secondary to an interstitial constitutional PHEX gene deletion. Her severe phenotype and multiple genotypic findings reinforce the importance of thorough genetic testing in the setting of complicated phenotypic presentations.
INTRODUCTION
The 2q37 locus is a commonly deleted subtelomeric regions (Supplemental Fig. 1).[1] A subset of patients, with a distal deletion, present phenotypic features which are similar to those found in Turner syndrome (TS) (Table 1). The 2q37 microdeletion syndrome is characterized by mild-moderate developmental delay, brachymetaphalangy,[2] short stature, obesity, hypotonia, characteristic facial appearance, seizures, congenital heart disease (coarctation/hypoplasia), hydrocephalus, dilated ventricles, genitourinary anomalies (primary gonadal failure), gastrointestinal abnormalities, and renal malformations (horseshoe kidney/renal dysplasia).[3] The clinical phenotype is quite variable (Table 1). Multiple skeletal anomalies have been described in patients with 2q37 deletion which overlap with those described in TS, including but not limited to: brachydactyly type E, scoliosis, brachymetacarpia, osteopenia, hypoplastic ulna, vertebral abnormalities, pectus excavatum, and bilateral dislocated hips with acetabular dysplasia.
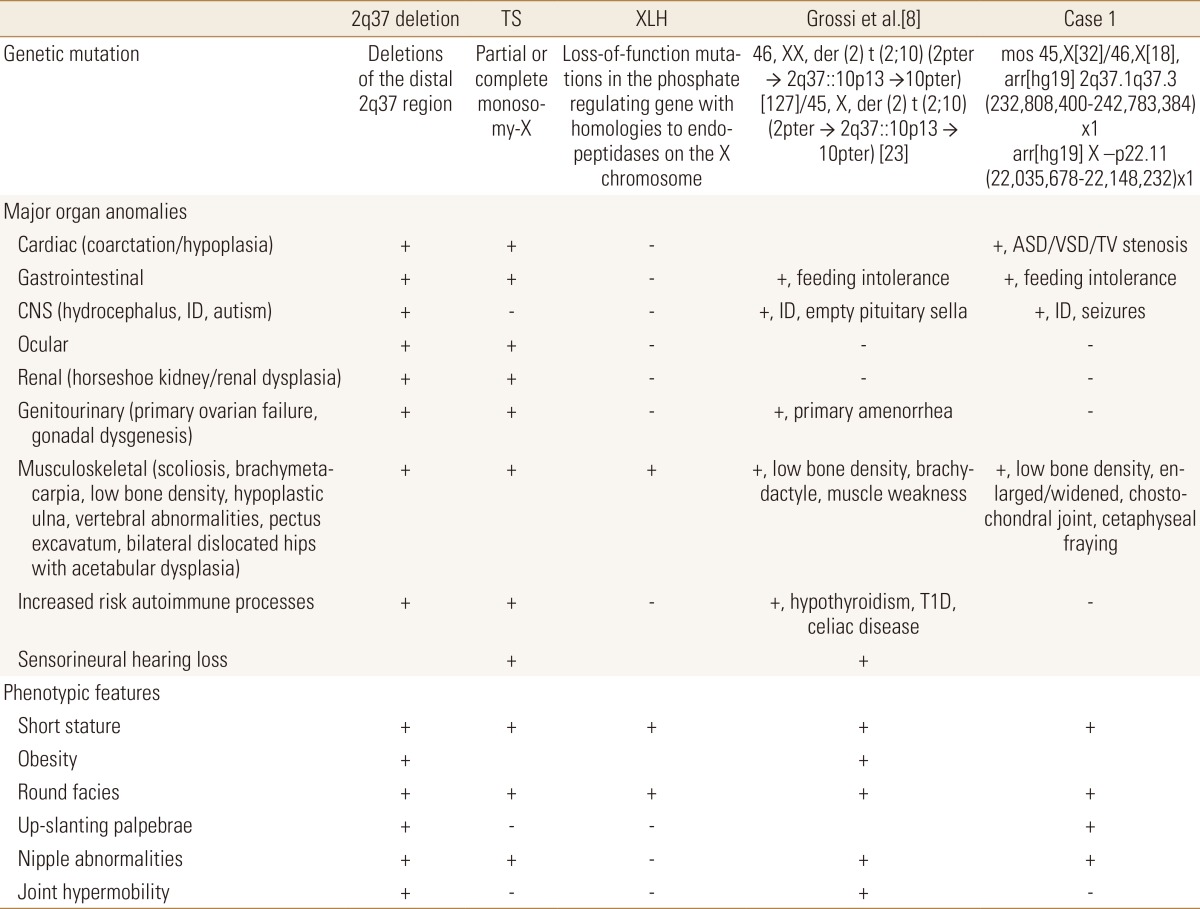
Genotypic and phenotypic findings of patients with 2q37 deletion syndrome, TS and XLH
To our knowledge, even though skeletal anomalies are common in both TS and 2q37 deletion syndromes, no patients have been reported with a 2q37 deletion with abnormal sex chromosome and clinical or biochemical features of X-linked hypophosphatemic rickets (XLH). XLH is an X-linked dominant disorder characterized by renal phosphate wasting, aberrant vitamin D metabolism, and abnormal bone mineralization with low serum phosphate, low to normal serum 1,25-dihydroxy-vitamin D, normal serum calcium and parathyroid hormone (PTH), and elevated alkaline phosphatase activity.[45] Loss-of-function mutations in the phosphate regulating endopeptidase homolog, X-linked (PHEX) have been causally associated with XLH.[6] The PHEX gene is located on the X chromosome, at Xp22.1-22.2 and spans 225 kb of genomic sequence.[7] One mutant PHEX gene is sufficient to result in the disease phenotype.[4] The phenotype although variable in its expressivity, is characterized by: short stature, bone pain, enthesopathy, and radiographic evidence of rickets, osteomalacia and lower extremity deformity.[67] Thorough genetic evaluation is required to ensure identification of concomitant deletion syndromes and aneuploidy to ensure appropriate diagnoses are made.
CASE
We report a 3-year-old female with a congenital heart defect, microcephaly (−3.23 standard deviations [SD]), developmental delay, dysmorphic features (prominent forehead, highly arched eyebrows, deep-set eyes, a flat nasal bridge, tented upper lip with a short philtrum), short stature (−4.13 SD) and poor weight gain (−2.75 SD) found to have XLH secondary to multiple chromosome anomalies including a small interstitial microdeletion of part of the PHEX gene in the setting of mosaic TS (mos 45,X[32]/46,X[18])(Supplemental Fig. 2). She was also found to have a 9.98 Mb terminal deletion of chromosome 2: del(2)(q37.1;q37.3) which explains her associated clinical findings mentioned above.
A 23-month-old female infant was admitted to the hospital for poor weight gain and was found to have rachitic changes on imaging associated with mild hypocalcemia and hypophosphatemia. Her growth parameters at birth were as follows: length −4 SD, weight −6 SD, head circumference −2.3 SD. She was born full term, with complicated delivery due to respiratory distress. On admission her growth parameters were as follows: length −5.4 SD, weight −10.4 SD, head circumference −3.2 SD. Physical exam revealed microcephaly, rachitic rosary palpable on costal margins, bowing of her upper extremities bilaterally and widening of her wrists bilaterally. No dental defects appreciated. Her vision and hearing were normal. Initial laboratory analysis revealed: phosphate 2.3 mg/dL (4.5–6), calcium 7.9 mg/dL (8–10.2), ionized calcium 4.3 mg/dL (4–7), PTH 401 pg/mL (15–65), alkaline phosphatase 530 U/L (80–270), 25-hydroxy-vitamin D 22 µg/L (>30), urine phosphate 270 mg/dL, tubular reabsorption of phosphate 40%, and fibroblast growth factor-23 494 RU/mL (<230). Skeletal survey revealed evidence of rickets including prominent beading/knobs of bone at the costochondral joint, osteopenia, fraying of bilateral humeri and femurs (Fig. 1).

Skeletal survey obtained at the time of diagnosis. Significant for severe diffuse osteopenia, with diffuse metaphyseal irregularities and flaring with multiple fractures.
Review of the karyotype obtained at 3 months of age revealed 50-cell analysis with 65% 45, X and 35% 46, XX (mos 45,X[32]/46,X[18]). Her karyotype was concerning for a cell line with a mosaic translocation between the X chromosome and chromosome 2, and a second cell line that was determined to have the same translocation with a loss of the derivative X chromosome. The Affymetrix CytoScan HD array (Affymetrix, Santa Clara, CA, USA) was used for her chromosome microarray analysis to confirm the abnormality. Chromosomal microarray analysis revealed a 9,975 kb microdeletion of the terminus of chromosome 2 at 2q37.1 and a 112.6 kb microdeletion at Xp22.11 (arr[hg19]2q37.1q37.3[232,808,400-242,783,384]×1; arr[hg19] Xp22.11 [22,035,678-22,148,232]×1). The microdeletion at Xp22.11 included the PHEX gene (Online Mendelian Inheritance in Man [OMIM] # 307800) which confirmed the diagnosis of XLH. Mosaic loss of the X chromosome was not detected by array. Two normal copies from the terminal short arm of the X chromosome up until the small 112.6 kb deletion and a terminal deletion of chromosome 2: del(2)(q37.1;q37.3) explained her additional clinical findings. She was started on oral supplementation with calcium, phosphate, calcitriol and cholecalciferol and her lab values improved: calcium 9.7 mg/dL, phosphate 3.6 mg/dL, PTH 64 pg/mL.
DISCUSSION
This is the first case report of a mosaic TS (mos 45,X[32]/46, X[18]), 2q37 deletion syndrome, del(2)(q37.1;q37.3), and XLH secondary to interstitial deletion of the PHEX gene. Given her variable phenotypic presentation, genetic testing was completed which revealed that she had three concurrent genetic diagnoses. The clinical phenotype for 2q37 deletion syndrome and TS overlap; however, the genotypic comparison is not well defined. Grossi et al.[8] described a case of a patient with mosaic TS with partial monosomy 2q and trisomy 10p associated with multi-organ autoimmunity. Per report, this patient presented with short stature, low bone density and brachydactyly. This patient had no imaging findings or biochemical parameters consistent with XHL (Table 1).[8] As is clearly outlined in Table 1 our patient and the patients described by Grossi et al.[8] share many similar phenotypic features including musculoskeletal abnormalities however the later showed no evidence of rickets.
The association of TS and X-linked dominant conditions, such as XLH, raises questions regarding the mechanistic and genetic etiologies of these phenotypic findings. It remains unclear whether these phenotypic findings are a result of haploinsufficiency or a dominant negative effect resulting in disease. After extensive review of the literature; no previous case reports have described a case of TS and XLH. Compared to other X-linked dominant disorders, in XLH, females and males are affected with equal severity. Studies in affected females indicate they have a normal X chromosome inactivation pattern. The presence of disease in affected females could be accounted for by either a dominant-negative effect or haploinsufficiency. Our patient has mosaic TS (mos 45,X[32]/46,X[18]) detected by karyotype. The microarray detected a PHEX deletion which occurred on the short arm of the X chromosome. Since microarray only evaluated the 46, XX genotype, the PHEX deletion must have occurred early on in development on one X chromosome of the 46, XX cells and therefore resulted in a severe phenotypic effect. The karyotype that was performed revealed the 45, X genotype which would not have been discovered if a comprehensive genetic evaluation was not completed. In addition, by definition, there are differing degrees of mosaicism in various tissues therefore impossible to determine the exact influence of the deletion on the organ level.
Ideally, further familial segregation studies and additional investigation would be beneficial. A karyotype and microarray from the parents would allow for evaluation of any structural abnormalities including a balanced translocation or to determine whether the PHEX deletion is inherited from either parent or a de-novo finding. Unfortunately, parental samples were not available from this family.
CONCLUSION
This is the first case report describing concurrent mosaic TS, 2q37 deletion syndrome, mos 45, X[32]/46,X[18], del(2)(q37.1;q37.3), and XLH. Apart from both TS and XLH being linked to the X chromosome, the long-term prognosis of individuals with both chromosomal anomalies is not known. We postulate that the deletion of the PHEX gene must have occurred early in development on one X chromosome of the 46, XX cells causing XLH, in our patient and could potentially have impact on the long term management needs of this patient. There is no evidence to suggest an association or causation among these three disorders; it most likely was a random occurrence.
Notes
No potential conflict of interest relevant to this article was reported.
References
SUPPLEMENTARY MATERIALS
Supplementary Fig. 1
Deletions of the distal 2q37 region involve the last cytogenetic band on the long arm of chromosome 2, which is divided into three subbands (2q37.1, 2q37.2, and 2q37.3). The last sub-band notably contains a small subtelomeric region, 2qtel, which displays non-deleterious polymorphic deletions or duplications.
Supplementary Fig. 2
Dysmorphic features present in our patient include: prominent forehead, highly arched eyebrows, deep-set eyes, a flat nasal bridge, and tented upper lip with a short philtrum.