INTRODUCTION
One of the most evolutionarily ancient and well-studied signaling pathways are the group of mitogen-activated protein kinases (MAPKs), which are present in all eukaryotes, including yeast, where MAPKs regulate responses to pheromones, cell integrity, high osmolarity, or other signals.[1,2] While MAPK pathways are ancient, they underwent a major diversification with the development of vertebrate life concurrent with the evolution of skeletal tissues.[3] Accordingly, each of the major MAPK pathways of the conventional group, the extracellular signal-regulated kinase (ERK), p38 and c-JUN N-terminal kinase (JNK) pathways, have been demonstrated to have a key function in promoting bone formation by both osteoblasts [4-8] and also in regulating bone resorption by osteoclasts.[9-13]
In this review, we focus on the contribution of the ERK pathway to osteoblast differentiation. Both ERK isoforms, ERK1 (MAPK3) and ERK2 (MAPK1) are expressed in osteoblast-lineage cells. The distal portion of the ERK signaling cascade displays a stereotypical “wiring” that is largely similar across cell types, with the MAP2Ks MAPK kinase (MEK)1 and MEK2 serving to activate ERK via phosphorylation. As indicated by mouse genetics, though there are differences in biochemical function, ERK1 and ERK2 have partially redundant roles.[14,15] Similar partial redundancy is seen with MEK1 and MEK2, which is most evident in the development of placental defects in either Mek1−/− or Mek1+/−Mek2+/− embryos.[16] Interestingly, a knock-in of the Mek2 protein-coding sequence into the Mek1 locus demonstrated that Mek-associated developmental phenotypes are driven by the total Mek1 and Mek2 gene dosage, with the MEK1 and MEK2 proteins able to substitute for one another. Upstream of MEK1 and MEK2 MAP2Ks, a wide range of kinases, large kinases in the MAP3K family, serve to activate the pathway in a manner that is both stimulus and cell type-specific, with the most classic activator of the ERK MAPK pathway being the RAF family of kinases. However, as discussed below, a wide range of MAP3Ks contribute to ERK activation in osteoblasts.
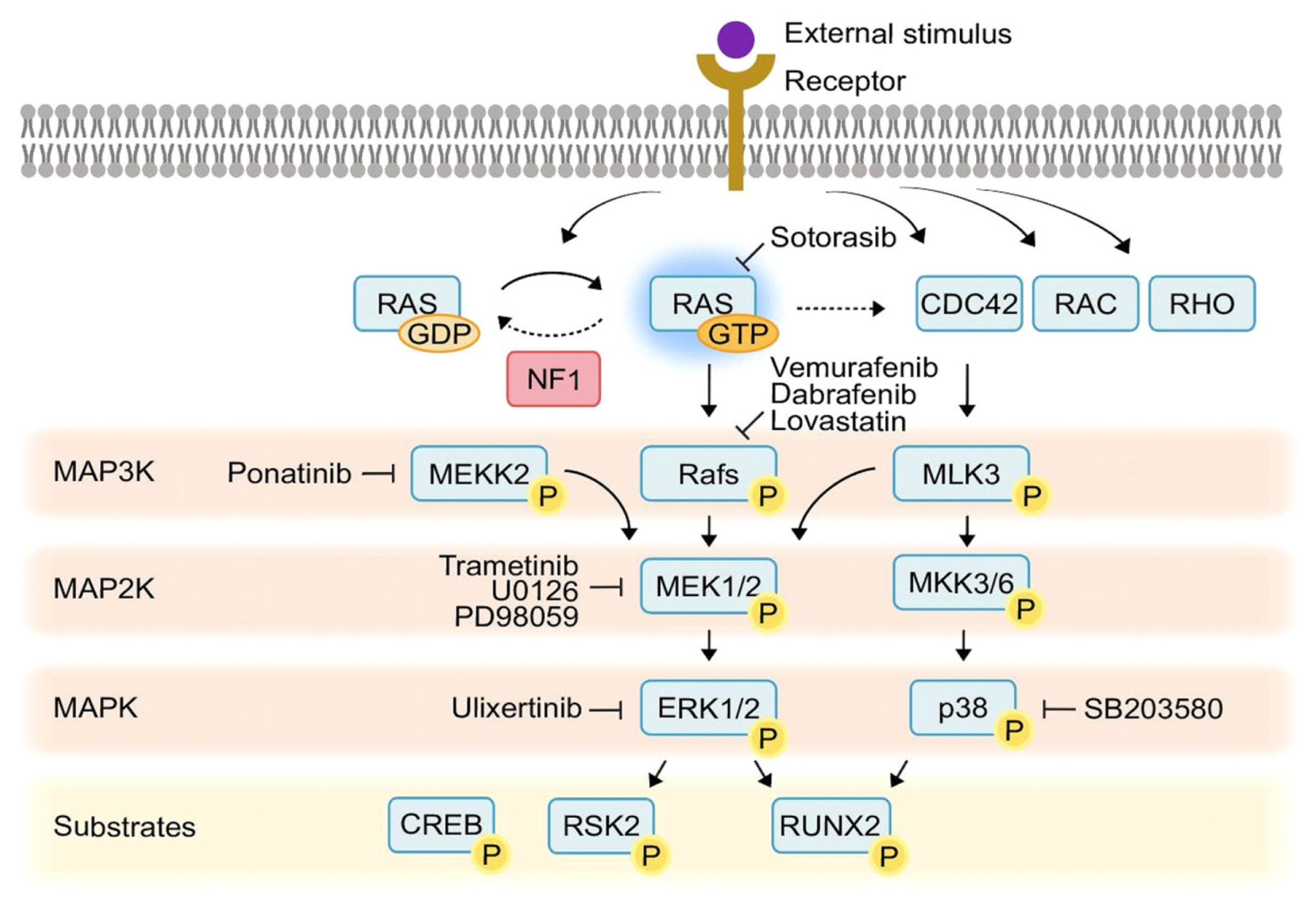
Collectively, mutations in the ERK MAPK pathways are the most common genetic lesions in cancer, and include common recurrent mutations in BRAF and KRAS seen across many tumor types, in addition to other less common mutations in ERK MAPK pathway components such as MEK1 or mutations in upstream signaling components, including EGFR that ultimately lead to ERK activation.[17] Accordingly, this critical importance of ERK in tumorigenesis has led to introduction of a wide range of ERK MAPK pathway inhibitors for treatment of a wide range of tumors, resulting in a powerful pharmacopeia to modulate the ERK MAPK pathway in patients. These drugs include inhibitors of MEK1 and MEK2 such as trametinib,[18] inhibitors of BRAF, a frequently mutated MAP3K upstream of ERK, including vemurafenib and dabrafenib,[19] or inhibitors of mutated KRAS, such as the recently approved sotorasib.[20,21] Due to this, an increasing number of patients are exposed to ERK MAPK pathway inhibitors, and the skeletal consequences of this inhibition must be considered,[22] especially as outcomes and life expectancy for these patients improve and raise the relative importance of long-term sequelae of ERK MAPK pathway inhibition. This development of ERK MAPK pathway inhibitors has also provided a series of potential therapies for skeletal disorders where ERK MAPK pathway overactivation may represent an important pathogenic mechanism, such as neurofibromatosis type 1 (NF1).[23,24] Thus, understanding the role of the ERK MAPK pathway in bone formation by osteoblasts is important both for the intrinsic importance of ERK MAPKs as major regulators of osteoblast differentiation and also due to the wealth of clinically-approved small molecule drugs targeting this pathway (Fig. 1).
PHYSIOLOGIC FUNCTION OF THE ERK MAPK PATHWAY IN OSTEOBLASTS
Most studies on loss-of-function of ERK MAPKs emphasize their critical roles in promoting early commitment and differentiation of skeletal progenitors to the osteoblast lineage and skeletal mineralization (Table 1). For instance, deletion of Erk1/2 in the limb bud mesenchyme and limb skeletal stem cells via Prx1-Cre results in severe hypo-mineralization of the limbs.[25] Similarly, deletion of Mek1/2 in early progenitors committed to the osteoblast lineage using osterix-Cre resulted in impaired bone formation,[8] and mice bearing a transgene of a dominant-negative MEK1 display hypo-mineralization of long bones and the calvarium.[6] While this clearly indicates a role for ERK MAPK in osteoblast differentiation or activity in vivo, somewhat complicating this picture is the observation that ERK MAPK pathway disruption in either Erk1−/−Erk2col2 or Erk1−/−Erk2Prx1 mice leads to defects in the growth plate, specifically an inability to clear and turnover hypertrophic chondrocytes.[25] Given evidence from several investigators that the growth plate cells undergo a trans-differentiation or plasticity process that contributes to the production of marrow-resident osteoblasts,[26-28] this raises the possibility that the blockade in hypertrophic chondrocyte clearance could in turn block the contribution of growth plate cells to the marrow osteoblast pool. Thus, ERK MAPKs could act not only to regulate the differentiation of committed osteoblast lineage cells, but also in pre-commitment progenitors, such as those within the growth plate, to regulate initial commitment to the osteoblast lineage. Notably, growth plate cells appear to only account for a portion of the osteoblasts present in long bones, thus ERK MAPKs may have separate but also important contributions in cathepsin k (CTSK)-lineage periosteal stem cells [29] or leptin receptor (LEPR)+/chemokine (C-X-C motif) ligand 12 (CXCL12)-abundant reticular cells [30-32] or other skeletal stem and progenitor populations.
Interestingly, recent work in a zebrafish model of osteoblast-mediated scale regeneration finds evidence that ERK activation propagates across the entire tissue in a synchronous wave that ultimately coordinates the regeneration process.[33] Evidence from a combination of modeling and proof-of-concept studies using transgenic fibroblast growth factor (FGF) family member expression suggests that this synchronized wave of osteoblast ERK activation is mediated by radial diffusion of an activating ligand, such as FGF, combined with induction of a refractory state in ERK activation after stimulation. This raises the possibility that, if this phenomenon is conserved, ERK MAPKs may not only promote osteoblast activity but mediate broader spatiotemporal coordination of bone formation. Thus, it will be of particular interest to apply a real-time reporter of ERK activity to the bone to examine tissue-wide coordination of skeletal ERK activity in a mammalian system.
LIGANDS ACTING UPSTREAM OF ERK MAPKS IN OSTEOBLASTS
A variety of stimuli activate the ERK MAPK pathway to coordinate signaling during osteoblast differentiation and skeletogenesis. FGF/FGF receptor (FGFR)-mediated signaling regulates osteoblast-lineage commitment, proliferation, maturation, and apoptosis.[34] Numerous in vitro studies demonstrated that the ERK MAPK pathway is required for FGFs-induced osteoblast differentiation through Runt-related transcription factor 2 (Runx2) phosphorylation and activation.[35,36] Genetically, ERK activation is closely linked to the presence of FGFR2 mutations (S252W or P253R) associated with human Apert syndrome.[37-39] Similar ERK activation has also been observed with the craniosynostosis-associated FGFR2 mutation (E731K).[40] A knock-in mouse model carrying a mutation encoding for the P253R alteration in Fgfr2 exhibited ERK activation and premature fusion of the coronal suture, a shortening of the cranial base and long bone growth plates.[37] ERK MAPK pathway inhibition with a MEK1/2 inhibitor partially reversed these phenotypes, including the premature closure of the coronal suture and the growth phenotype in long bones, with these results matching mechanistic studies demonstrating that FGFR2 activation engages the ERK pathway to phosphorylate and stabilize Runx2 via a Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (PIN1)-dependent mechanism.[39,41-44]
Insulin-like growth factor 1 (IGF-1) regulates skeletal homeostasis via autocrine and paracrine signaling.[45] Intensive studies showed bone anabolic effects of IGF-1/IGF-1 receptor (IGF-1R) signaling. IGF-1 stimulates mesenchymal stem cell (MSC) proliferation and osteogenic differentiation, and mineralization capacity. Accordingly, Igf-1 overexpression in osteoblasts increased trabecular bone density,[46] whereas IGF-1R deletion in osteoprogenitor cells (Igf1rOsx) or mature osteoblasts (Igf1rOcn) caused delayed mineralization and reduced bone formation.[47,48] ERK activation is required for IGF-1-induced proliferation and osterix expression in human MSCs,[49,50] yet the role of the ERK MAPK pathway in mediating the osteoanabolic effects of IGF-1 signaling in vivo requires further study.
The wingless-type MMTV integration site family member (WNT) signaling pathway also activates ERK MAPK to exert its bone anabolic effect in osteoblast-lineage cells.[47,51] WNT signaling is subdivided into canonical (β-catenin-dependent) and noncanonical (β-catenin-independent) pathways and noncanonical pathway includes MAPK activation, though emerging evidence indicates that the ERK MAPK pathway can also impact canonical signaling. Both in vitro and in in vivo studies showed that WNT3a activates ERK MAPK to induce osteoblast precursor proliferation and differentiation.[52] Likewise, ERK activation suppresses the activity of glycogen synthase kinase 3β, an upstream inhibitor of β-catenin, resulting in bone formation.[51] In addition, mice lacking the WNT receptor Frizzled-9 showed low bone mass without alteration in the canonical WNT/β-catenin signaling pathway, and ERK activation mediated by WNT5a is required for the bone anabolic effect of Frizzled-9 stimulation.[53]
Other upstream stimulators of the ERK MAPK pathway in osteoblasts include the transforming growth factor-β (TGF-β) superfamily, including bone morphogenetic protein (BMP) ligands. These transduce signals to ERK MAPK via the non-SMAD-dependent pathway to regulate stem cell differentiation during bone formation and bone homeostasis.[54] Deletion of TGF-β type I receptor (Alk5) in skeletal progenitors by Dermo1-Cre revealed that TGF-β promotes osteoblast proliferation and early differentiation through a combination of both the SMAD2/3 and ERK MAPK signaling pathways.[55] At the cellular level, TGF-β induces activator protein-1 (AP-1) activation ERK activation.[56] ERK signaling mediates BMP2 or 7-induced osteogenesis and Runx2 expression, whereas constitutive activation of ERK signaling decreased BMP2-induced Runx2 expression and activation.[57,58] BMP2 stimulation leads to Runx2 acetylation and stabilization, potentiating Runx2 transcriptional activity.[59] However, further study is needed to clarify the degree to which the ERK pathway specifically accounts for the activity of BMP stimulation during skeletogenesis.
MAP3KS ACTIVATING THE ERK MAPK PATHWAY IN OSTEOBLASTS
While RAF isoforms (ARAF, BRAF, and c-RAF/RAF1) are classical MAP3Ks that serve to activate MEK1 and MEK2 MAP2Ks, particularly in a variety of oncologic contexts, it is clear that a broader diversity of MAP3Ks function upstream of ERK MAPKs in osteoblasts, often in combination with concurrently activating other MAPK pathways. In particular, studies of BRAF using a Col2-Cre line that targets a wide range of skeletal cells find that the knockout mice have grossly normal initial skeletogenesis.[60] Similarly, deletion of Braf with the osteoblast-targeting Col1-Cre did not produce histologically evident changes in bone architecture in 2-week old mice. Mice with a conditional deletion of c-Raf/Raf1 using Col2-Cre have also been examined, finding impaired clearance of hypertrophic chondrocytes in the growth plate, a finding that may be expected to alter the production of growth-plate derived osteoblasts, though this has yet to be examined.[61] It will be of particular interest to expand and update these findings to see whether RAF isoforms have a more subtle role in regulation of adult bone homeostasis and whether redundancy might be masking the contribution of RAF to osteoblast function.
Given this negative initial data with RAF isoforms, there has been interest in identifying other MAP3Ks that serve to activate ERK MAPKs in osteoblasts. MLK3 (MAP3K11) serves to activate both p38 and ERK downstream of BMP stimulation, and activation of MLK3 induces Runx2 phosphorylation at a combination of p38- and ERK-mediated sites.[62] MLK3 acts downstream of the guanosine triphosphatase (GTPase) CDC42, binding through a CDC42/Rac interactive binding (CRIB) region in MLK3. Accordingly, mutation of the CRIB region phenocopies the skeletal abnormalities of an MLK3 null allele. CDC42 is itself activated by the GEF faciogenital dysplasia 1 (FGD1).[63] As FGD1 is mutated in the human skeletal dysplasia disorder FGD, defective MLK3 signaling is likely a key pathogenic mechanism for this disorder. This also provides a clear example of how impaired ERK signaling in osteoblasts can serve as a basis for human skeletal diseases.
MEKK2 (MAP3K2) is a member of the MAP3K family, and early in vitro biochemical studies have shown that MEKK2 has the capacity to activate downstream MAPK pathways including ERK1/2, JNK, p38, and ERK5.[64-67] Studies of MEKK2 in osteoblasts demonstrated that MEKK2 promotes osteoblast activity by both mediating a nonclassical pathway for deubiquitination and stabilization of β-catenin.[68] MEKK2 has also been suggested to regulate the activation of the JNK pathway downstream of SMURF1 loss-of-function in osteoblasts.[69] However, later studies in MEKK2 deficient osteoblasts failed to find a clear contribution to JNK activation, at least under basal conditions, so the contribution of SMURF1 regulation of MEKK2 activity to osteoblast differentiation and activity remains unclear.[4,68] In keeping with these important functions of MEKK2, MEKK2-deficient mice display severe cortical and trabecular osteopenia alongside calvarial hypomineralization.[68] Recent findings additionally demonstrate that MEKK2 is a key mediator of the aberrant ERK activation occurring in osteoblasts downstream of loss of function of NF1, a GTPase-activating protein that negatively regulates RAS activation.[70] Despite MEKK2-deficient mice displaying severe osteopenia, either genetic or pharmacologic inhibition of MEKK2 prevented the constitutive ERK activation occurring in models of skeletal NF1 disease and overall ameliorated the NF1-associated skeletal phenotypes. This work, alongside several prior studies establishing the ERK MAPK pathway as one, though likely not the only, mediator of NF1-associated skeletal diseases,[23,24,71] argues that ERK MAPKs are not unconditionally pro-anabolic. Rather, there is likely a window of activity that is pro-anabolic, with degrees of activation outside of this range either not promoting osteoblast differentiation or having a deleterious effect. This “window effect” may also have an etiology in the distinct sets of downstream substrates for the ERK MAPK pathway that appear in early versus late-stage osteoblasts, with overactivation of ERK perhaps prolonging ERK activity during the differentiation process and engaging distinct sets of effectors in mature osteoblasts. Ultimately, further study on the context-dependent roles of the ERK MAPK pathway will be needed to determine the basis for this apparent window in ERK activity that allows for bone anabolism. Additionally, a comparison of these studies on MEKK2 and MLK3 emphasizes how each function in a distinct biochemical context, functioning downstream of different upstream activators in a selective manner. This reinforces the general concept that MAP3Ks serve as key integration points that activate downstream MAPK pathways in a manner that is context and cell-type specific. This specificity also suggests that MAP3Ks are a promising point to modulate ERK MAPK signaling while also minimizing the toxicity associated with broad alterations of this essential pathway across multiple tissues.
REGULATORS OF THE ERK MAPK PATHWAY IN OSTEOBLASTS
The NF1 protein negatively regulates RAS MAPK pathway through its GTPase activity. Loss of function NF1 mutations in humans causes NF1, a prevalent genetic disorder with an incidence of approximately 1/3500.[72] NF1 syndrome is characterized by cutaneous neurofibromas and neural tumors, but also a wide variety of non-tumor manifestations including skeletal dysplasia.[73,74] Patients with NF1 display skeletal abnormalities including craniofacial dysmorphogenesis, osteopenia/osteoporosis, short stature, and impaired fracture healing.[75,76] Similarly, conditional mouse models lacking NF1 in various skeletal lineage cells display characteristic features of the skeletal abnormalities of NF1 patients, including impaired bone structure and severe cortical bone porosity in Nf1Prx1,[77] Nf1Col,[71] Nf1Osx,[78] and Nf1Dmp1,[70] mice. To understand the pathogenesis of NF1 skeletal manifestations, several studies have investigated target pathways mediating the aberrant ERK effects of NF1 loss of function in skeletal lineage cells. It has been reported that RAS inhibition by lovastatin attenuates the constitutive activation of ERK MAPKs in osteoblasts isolated from Nf1Col2 mice.[71] NF1-deficiency in bone-forming cells causes a RAS/ERK-dependent increase in the endogenous inhibitor of mineralization, inorganic pyrophosphate, ultimately leading to a bone mineralization deficit.[78]
Interestingly, altered ERK MAPK pathway regulation can result in divergent skeletal phenotypes. Protein-tyrosine phosphatase, nonreceptor-type 11 (PTPN11), also known as SHP2, is a SH2 domain-containing non-receptor protein tyrosine phosphatase that acts to regulate ERK activity in cell type and context-dependent manner, particularly downstream of receptor tyrosine kinase stimulation.[79] As with NF1, while ERK is a prominent downstream mediator of PTPN11 it is not the only downstream target, and other pathways, such as nuclear factor of activated T cells calcium signaling or PKA signaling can also be regulated.[80,81] Gain-of-function mutations in PTPN11 enhance ERK signaling in osteoblasts and are seen in approximately 50% of patients with Noonan syndrome, which is associated with skeletal manifestations including impaired skeletal mineralization with onset in childhood and short stature.[82-84] Interestingly, loss of function in PTPN11 is associated with a distinct metachondromatosis phenotype including cartilaginous proliferation throughout the skeleton in both humans and mice.[85,86] This activity of PTPN11 has been mapped to its function in early skeletal stem cells, including a specific periosteal stem cell expressing CTSK and other markers.[29,80,85] The action of PTPN11 in early skeletal stem and progenitor cells has been associated with alterations in lineage selection, both skewing osteoblast versus chondrocyte lineage towards chondrocytes by regulating Sox9 activity and also inhibiting the trans-differentiation of terminal growth plate chondrocytes into osteoblasts.[80,87] These functions of PTPN11 are context-dependent, as deletion of PTPN11 in more mature, Bglap-expressing osteoblast lineage cells led to a very distinct phenotype of severe osteopenia.[88] Consistent with the known substrates of the ERK MAPK pathway in osteoblasts discussed below, this phenotype was associated with regulation of Runx2 transcriptional activity.
Lastly, another regulatory element of MAPK signaling active in osteoblasts is cross-regulation by distinct MAPK pathways.[89] For instance, loss of function in the JNK pathway, including loss of function in MAP3Ks upstream of JNK such as TAOK3 (MAP3K18), produces a marked increase in ERK activation.[90] Likewise, the widely utilized p38 inhibitor SB203580 can upregulate ERK activity in osteoblasts,[91] though this may specifically reflect the ability of SB203580 to induce MLK3 activity.[92] Thus, MAPK pathways may cross-regulate, and this cross-regulation may serve as a mechanism for signal integration in osteoblasts.
ERK MAPK SUBSTRATES IN OSTEOBLASTS
Runx2 emerged as one of the first targets of ERK MAPKs in osteoblasts in a series of biochemical studies finding that ERK activity modulated the ability of Runx2 to transactivate an osteocalcin promoter fragment through the ability of ERK to phosphorylate Runx2.[35,93] Later biochemical analysis identified ERK phosphorylation at S43, S301, S319, and S410, with dual mutation of the S201 and S319 sites rendering Runx2 refractory to MAPK induced functional activation.[36] Biochemically, Runx2 interacts with ERK MAPKs via a classical MAPK-binding D-domain in the protein/serine/threonine-rich domain in the C-terminus.[35,94] Phosphorylation of Runx2 by ERK partially overlaps with phosphorylation by p38, with both kinase families targeting the S319 site in particular.[5,94] This ERK-mediated Runx2 activation has been mapped to acting downstream of FGF stimulation, with ERK-mediated phosphorylation of Runx2 at S301 leading to Runx2 stabilization and increased transcriptional activity in this context.[43] This effect is mediated at least in part by PIN1 being recruited to phosphorylated Runx2 by ERK MAPK, leading to Runx2 prolyl isomerization that in turn leads to Runx2 acetylation and stabilization.[41,95] Accordingly, Pin1-deficient mice display calvarial hypomineralization and clavicular hypoplasia, stigmata associated with reduced Runx2 activity.[96]
Mouse genetic studies from a variety of investigators have reinforced this link between ERK MAPKs and Runx2 by finding that mice with loss of ERK MAPK pathway function display a cleidocranial dysplasia-like phenotype, the combination of calvarial hypo-mineralization, clavicular hypoplasia, stigmata associated with haploinsufficiency of Runx2 in both humans and mice.[97,98] Similarly, deletion of Mek1/2 using osterix-Cre produces a cleidocranial dysplasia-like phenotype.[8] Conversely, several lines of evidence suggest that a gain in ERK activity leading to increased Runx2 activity contributes to craniosynostosis pathogenesis, particularly in models of Apert syndrome associated with FGFR2 gain-of-function mutations. For instance, ERK inhibitors can block cranial suture closure associated with FGF2 stimulation.[44] Mechanistically, FGF stimulation leads to Runx2 phosphorylation via the ERK MAPK pathway and subsequent PIN1-dependent Runx2 acetylation and stabilization, ultimately increasing Runx2 transcriptional activity.[42,43] Thus, craniosynostosis and cleidocranial dysplasia can be associated with ERK pathway gain-of-function and loss-of-function, respectively, with Runx2 serving in both cases as a key downstream target. This offers in vivo phenotypic validation of the importance of the ERK MAPK pathway for Runx2 activity and raises the possibility that FGFR2 mutation-associated craniosynostosis and cleidocranial dysplasia phenotypes can be considered as opposite ends of a phenotypic continuum of ERK and downstream Runx2 activity in osteoblasts.
Another important ERK MAPK substrate is the AGC kinase p90 ribosomal S6 kinase (RSK2), where ERK activates RSK2 via phosphorylation.[99] Phosphorylated RSK2 in turn phosphorylates and activates activating transcription factor 4 (ATF4), a driver of collagen transcription in mature osteoblasts.[23] Accordingly, mice and humans lacking Rsk2 show low bone mass due to impaired production of type I collagen, the major organic component of bone, with RSK2 mutations in particular associated with Coffin-Lowry syndrome, which includes prominent skeletal dysplasia features.[100] Considered together, it is particularly interesting that 2 prominent ERK MAPK substrates in osteoblasts, Runx2 and ATF4, have functions that appear to parse respectively to the early and the late stages of osteoblast differentiation. This raises the possibility that the ERK MAPK pathway could have quite distinct functions in early versus mature osteoblasts, as these functional differences could partition based on downstream substrate expression or function.
While these are the best-studied effectors of ERK function in osteoblasts, there are likely many other relevant ERK MAPK substrates, including the Fos family (Fos, Fra1, Fra2, Fosb) of subunits of the AP-1 transcriptional complex. While subunits such as FOS or FRA1 can be phosphorylated and stabilized by ERK and also play important roles in osteoblast biology,[101-103] it remains unclear the degree to which ERK activation is specifically driving FOS and AP-1 activity in osteoblasts. In particular, mice with a knock-in mutation in the ERK phosphorylation site of FOS have intact osteoblast parameters, calling into question whether FOS is a relevant ERK effector in osteoblasts.[104]
THE ERK5 MAPK PATHWAY IN OSTEOBLASTS
Alongside, ERK1/2, p38, and JNK isoforms, ERK5 is also a member of the so-called “conventional” group of MAPKs, which are defined by the presence of a Thr-Xaa-Thr motif in the activating loop.[105] ERK5 is activated by the upstream MAP2K MEK5, and this pathway is intact in osteoblast lineage cells. One notable feature of ERK5 is its relatively high degree of homology with ERK2 and the presence of a threonine-glutamate-tyrosine motif in the activation loop identical to that in ERK1 and ERK2.[106] Similarly, MEK5 displays homology with MEK1 and MEK2 and is also targeted by several of the most commonly used MEK1/2 inhibitors, including U0126 and PD98059, raising the possibility that the MEK5/ERK5 pathway may account for some of the activity ascribed to the ERK1/2 pathway in studies using MEK inhibitors.[107] Despite these similarities, mouse genetic loss of function studies established that the ERK1/2 and ERK5 MAPK pathways have distinct functions in vivo.[108]
The function of the ERK5 MAPK pathway in osteoblasts remains controversial due to conflicting results using chemical inhibitors or in vitro systems. For instance, in vitro studies with cell lines and an ERK5 inhibitor show that ERK5 inhibition reduced osteoblast differentiation downstream of fluid flow shear stress.[109] In contrast, treatment of mice with a MEK5 inhibitor in vivo results in increased osteoblast differentiation and bone formation.[110] It is possible that this discrepancy reflects either differentiation-stage or context-specific differences in ERK5 function or simply limitations of the small molecule inhibitors or culture systems used.
In vivo studies emphasize that the functions of ERK5 in bone are complex and context dependent. Conditional deletion of ERK5 using an Nkx3.1-Cre resulted in spinal deformity similar to scoliosis and trabecular osteopenia in vertebral bodies.[111] However, histomorphometry studies on these mice demonstrated a hyperdynamic state with increases in both osteoclast and osteoblast activity associated with increases in the expression of the osteoclastogenic cytokine receptor activator of nuclear factor-κB ligand (RANKL) by osteoblasts and decreases in the RANKL decoy receptor osteoprotegerin. Thus, in this context, ERK5 regulates osteoclast differentiation via osteoblast-osteoclast coupling. Another study deleted ERK5 in early limb bud progenitors/stem cells using Prx1-Cre, finding delayed mineralization of long bones.[112] Mechanistically, this was linked with ERK5 phosphorylation of the SMAD1 linker region at S206 and SMURF2 at T249, which ultimately resulted in SMAD degradation, and ERK5-mediated suppression of SMAD-dependent Sox9 transcription. Accordingly, additional heterozygosity for Sox9 rescued the Erk5Prx1 phenotype of delayed mineralization. These results indicate an important role for ERK5 in skeletal mineralization, but leave open the question of to what degree this reflects a dysfunction in chondrocytes, osteoblasts, or early progenitors prior to commitment to these lineages. It will be of particular interest to deconvolute the contribution of these cell types using additional lineage-specific Cre lines and also to examine the contribution of ERK5 beyond the initial onset of mineralization to the maintenance of bone mass in older animals.
CONCLUSION
Given that a recurring theme in studies of the ERK MAPK pathway in the bone in that ERK function is highly dependent on cellular context, going forward, it will be important for our understanding of the ERK MAPK pathway to be updated in terms of our rapidly advancing understanding of the cell types comprising bone. Whereas a decade or more ago, the cells comprising bone were largely categorized in terms of simple morphologic and functional characteristics into chondrocytes, osteoblasts, and osteocytes, a much richer picture has emerged in recent work of many specific cell types and lineages that are discriminated through cell surface markers or genetic lineage reporters. These cell types include PTHrP+ growth plate-resident stem cells,[26] specific periosteal stem cells,[29,85] FACS defined skeletal stem cells that include populations both overlapping and not overlapping with these 2 former cell types,[113-115] ACTA2-lineage osteoprogenitors,[116] and multiple subsets of CXCL12-abundant reticular cells that comprise the marrow stroma,[30,31] many of which also express LEPR.[32,117] This discovery of an increasingly diversifying set of skeletal cell types is the important challenge that it is no longer sufficient to simply define how a given signaling pathway augments or inhibits osteoblast differentiation. Rather, it is now increasingly necessary to consider that the function of these pathways may differ substantially in each of these cell types, and that comparative genetic studies, likely largely using conditional knockouts targeted to several of these skeletal lineages, will be necessary to deconvolute the differential function of major signaling pathways to these cell types. This possibility for distinct functions in distinct cell lineages may at least partially underlie the commonly contradictory results obtained when signaling pathway functions are probed using cultures of mixed bone marrow stromal cells that are heterogeneous and contain several of these cell types. Given the robust function of the ERK MAPK pathway in regulating osteoblast differentiation, it is an outstanding candidate to serve as an initial template for how signaling pathways can serve starkly different functions in different specific skeletal populations.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print